
A mechanistic study on tumour spheroid formation in thermosensitive hydrogels: experiments and mathematical modelling†
Abstract
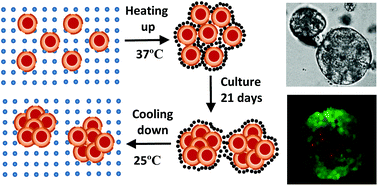
A tumour is a complex, growing tissue with a dynamic microenvironment. Its shape and size affect mass transport and thus the ability of drugs to penetrate into the tumour. Three-dimensional (3D) tumour spheroid culture has thus been recognised as an advanced tool for anti-cancer drug screening. However, the use of tumour spheroids has been hampered by the large variations in spheroid size, their irregular shape and the labour-intensive culture process. We explore thermosensitive hydrogels, P(NIPAM-AA), for culturing tumour spheroids and compare our approach with a traditional suspension culture method (non-adhesive surface) in terms of cell proliferation, tumour spheroid size distribution and spheroid morphology. Spheroids cultured in the microgel network show a narrower size distribution and a more spherical shape. We hypothesised that these observations could be explained by the fact that cells are homogeneously retained inside the hydrogels, cell aggregate formation is much slower due to network resistance and the cell death rate is smaller in comparison with the suspension culture. We developed a cellular automata (CA) model to validate these hypotheses. Spheroid formation with different parameter values, representing culture in suspension and in microgels, is simulated. Our results are consistent with the hypothesis that the microgel culture produces a more uniform size distribution of spheroids as a result of reduced cell death and the gel network resistance.
 Please wait while we load your content...
Please wait while we load your content...

