DOI:
10.1039/C6RA11592F
(Paper)
RSC Adv., 2016,
6, 70460-70467
Highly efficient dye-sensitized solar cells based on low concentration organic thiolate/disulfide redox couples†
Received
4th May 2016
, Accepted 19th July 2016
First published on 20th July 2016
Abstract
An imidazolium thiopropionate-based redox couple is introduced as an improved organic alternative to iodide/triiodide (I−/I3−) couples for dye-sensitized solar cells (DSSCs). The synthesized thiolate/disulfide (BMIT/BMIDT) redox couple shows negligible absorption in the visible spectral range, and higher redox potential (+0.72 V vs. NHE) compared with that of I−/I3−. With the optimized redox couple (such as 0.1 M/0.1 M)/dye N719 formulation, the fabricated DSSCs exhibit an average overall power conversion efficiency (PCE) of 6.8% and 8.1% under the simulated air mass 1.5 solar spectrum illumination at 100 and 50 mW cm−2, respectively. However, a higher concentration of the BMIT/BMIDT redox couple (such as 0.4 M/0.4 M) decreases the PCE of the devices, due to the desorption of dye molecules from the TiO2 photoelectrode surface.
Introduction
Dye-sensitized solar cells (DSSCs) have attracted both academic and industrial interest because of their high PCE, potential low-cost and easy-handling fabrication.1,2 A PCE up to 11.7% has been recently achieved using a traditional iodide/triiodide (I−/I3−) redox couple as a redox mediator.3 However, the sublimation of iodine and the absorption of visible light have limited the application of I−/I3− redox couples.4 Furthermore, the mismatch between the oxidation potentials of the I−/I3− redox couple and associated sensitizing dyes often limits the open circuit voltage (Voc) to 0.7–0.8 V.5 Therefore, the development of iodine-free redox couples is needed for large scale fabrication and commercial application of DSSCs.6
Recently, metal complexes,7–11 hole conductors,12–14 p-type semiconductors,15–19 Br−/Br3−,20,21 SCN−/(SCN)2,22,23 hydroquinone (HQ)/benzoquinone (BQ)24,25 and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) based redox shuttles have been synthesized and used as efficient redox mediators for DSSCs.26–29 However, most of these redox couples (except CoII/III) show relatively lower efficiencies than that of I−/I3−, due to relatively rapid electron recombination between photo-injected electrons in TiO2 photoelectrodes and the oxidized form of the redox couple in the electrolyte and/or slower dye regeneration.30 Recently, thiolate/disulfide based organic redox mediators have been extensively studied.31–34 For example, a 5-mercapto-1-methyltetrazole derived thiolate/disulfide (0.4 M/0.4 M) redox couple has achieved a PCE of 6.44% under standard AM 1.5 illumination at 100 mW cm−2.35 An efficiency of 4.0% was obtained for a thiolate/disulfide (0.2 M/0.2 M) organic redox couple with a metal-free organic dye.36 These results inspired us to design and synthesize a new type of organic redox couple for high PCE.
A thiopropionic acid-based disulfide/thiolate redox couple, BMIT/BMIDT, was synthesized and used for an iodine-free liquid electrolyte DSSC (see Scheme 1). This BMIT/BMIDT shows a higher redox potential than does the I−/I3− redox couple and it has negligible absorption in the visible spectral range. This negligible absorption results in high Voc and improved Jsc under load. The effect of the BMIT/BMIDT ratio, and the concentration effect of redox couples on the photovoltaic properties of these devices were further studied as well. A relatively low concentration of BMIT/BMIDT (0.1 M/0.1 M) with dye N719 [cis-bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)ruthenium(II)] as the photo-sensitizer exhibited a higher PCE than that of I−/I3− (0.1 M/0.1 M). However, higher concentrations of BMIT/BMIDT (0.4 M/0.4 M) yielded lower power conversion efficiencies due to desorption of dye from the TiO2 photoelectrode.
 |
| | Scheme 1 The synthetic route and chemical structures of BMIT and BMIDT. (a) (i) MeOH, KOH, 30 °C, 12 h, (ii) BMIBr, 12 h; and (b) (i) MeOH, KOH, 30 °C, 12 h, (ii) I2, 2 h, (iii) BMIBr, 12 h. | |
Experimental
Materials
Iodine, KOH, TiCl4 and H2PtCl6 were purchased from Sinopharm Chemical Reagent Co. Ltd. N-Methylimidazole, bromobutane, methanol, dichloromethane, and ethyl ether were purchased from Shanghai Chemical Reagents Co. (Shanghai, China). 4-tert-Butylpyridine (TBP) and thiopropionic acid were purchased from Aladdin Industrial Corporation (Shanghai). cis-Bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)ruthenium(II) (N719) was purchased from Solaronix SA (Switzerland). Fluorine-doped tin oxide overlayer (FTO) glass electrodes (7 Ω sq.−1), slurries containing 20 nm-diameter mesoporous and 200 nm-diameter light-scattering TiO2 were purchased from Dalian Hepat Chroma Solar Tech. Co. Ltd. 1-Butyl-3-methylimidazolium iodide (BMII) and 1-butyl-3-methylimidazolium bromine (BMIBr) were synthesized as earlier reported literatures.37
Synthesis of BMIT
In a methanol solution (20 ml), 3.17 g KOH (56.52 mmol) and 3.0 g (28.26 mmol) thiopropionic acid were mixed and stirred for 12 h to form a neutral solution at room temperature (RT). Then, 12.38 g (56.52 mmol) BMIBr was added into the solution. This mixture was stirred for 12 h, and we used rotary evaporation to remove the solvent. The precipitate was washed with dichloromethane and separated by filtration. The filtrate was washed with ethyl ether for three times. The final product was dried to give a yellow viscous liquid at 60 °C under vacuum. 1H NMR (400 MHz, D2O): 8.67 (s, 2H), 7.43 (s, 2H), 7.38 (s, 2H), 4.17–4.13 (m, 4H), 3.85–3.82 (m, 6H), 2.91–2.78 (m, 2H), 2.52–2.47 (m, 2H), 1.84–1.79 (m, 4H), 1.32–1.20 (m, 4H), 0.90–0.84 (t, 6H). 13C NMR (400 MHz, D2O): C14 (180.44), C4 (135.74), C1 (120.34), C2 (122.08), C7 (49.17), C6 (37.04), C8 (35.52), C11 (31.17), C9 (18.67), C10 (14.03), C12 (12.54), and MS (peak: 383.2433 (1+) can be found) were also carried out to confirm the existence of BMIT (Fig. S1†).
Synthesis of BMIDT
In a methanol solution (20 ml), 3.17 g KOH (56.52 mmol) and 3.0 g (28.26 mmol) thiopropionic acid were stirred for 12 h to form a neutral solution at RT. Then 3.58 g (14.13 mmol) iodine was added into the solution and sonicated for about 2 h until the iodine disappeared completely. The precipitate was removed by filtration. Then 6.19 g (28.26 mmol) BMIBr was added into the filtrate. This mixture was stirred for 12 h, and we used rotary evaporation to remove the solvent. The precipitate was washed with dichloromethane and separated by filtration. The filtrate was washed with ethyl ether three times. The final product was dried at 60 °C under vacuum to give a canary yellow viscous liquid. 1H NMR (400 MHz, D2O): 8.67 (s, 2H), 7.43 (s, 2H), 7.38 (s, 2H), 4.17–4.13 (m, 4H), 3.85–3.82 (m, 6H), 2.91–2.78 (m, 4H), 2.52–2.47 (m, 4H), 1.84–1.79 (m, 4H), 1.32–1.20 (m, 4H), 0.90–0.84 (t, 6H). 13C NMR (400 MHz, D2O): C17 and C20 (179.82), C4 (135.77), C1 (123.42), C2 (122.17), C7 (49.23), C6 (36.26), C11 and C16 (35.65), C12 and C15 (31.21), C9 (18.71), C10 (12.60), and MS (peak: 487.2436 (1+) can be found) were also carried out to confirm the existence of BMIDT (Fig. S1†).
Preparation of electrolytes
The compositions of the electrolytes used in this work are listed in Tables 1 and S1.†
Table 1 Electrolyte compositions for various cells examined in this work
| Electrolyte |
Composition |
| A |
0.1 M BMIT, 0.2 M BMIDT, 0.5 M TBP in acetonitrile |
| B |
0.1 M BMIT, 0.1 M BMIDT, 0.5 M TBP in acetonitrile |
| C |
0.1 M BMIT, 0.05 M BMIDT, 0.5 M TBP in acetonitrile |
| D |
0.1 M BMIT, 0.02 M BMIDT, 0.5 M TBP in acetonitrile |
| E |
0.1 M BMIT, 0.01 M BMIDT, 0.5 M TBP in acetonitrile |
| F |
0.1 M BMII, 0.1 M I2, 0.5 M TBP in acetonitrile |
| G |
1 M BMII, 0.1 M I2, 0.5 M TBP in acetonitrile |
Fabrication of DSSCs
The DSSCs were assembled following the previous reports.38 Two parallel edges with an adhesive tape was used to cover the cleaned FTO glass to control the thickness of mesoporous TiO2 film. Then two layers of TiO2 particles were followed depositing onto the FTO glass for further using. A 20 nm sized TiO2 particles with 10 μm thick was deposited onto the FTO glass through a doctor-blade technique. Then the FTO glass was dried at 115 °C for 5 min. A paste for the scattering layer containing 200 nm sized anatase TiO2 particles were coated on the top of the first TiO2 layer. The photoanodes were again gradually heated under an air flow at 125 °C for 5 min, 325 °C for 5 min, at 375 °C for 5 min, at 450 °C for 15 min, and at 500 °C for 15 min. The photoanodes were treated again by TiCl4 at 70 °C for 30 min and sintered at 500 °C for 30 min. After cooling to 80 °C, the TiO2 electrodes were immersed in 0.5 mM N719 acetonitrile and tert-butyl alcohol solution to complete sensitizer loading at RT for about 24 h. The dye-sensitized TiO2 electrodes were then washed with anhydrous ethanol and dried in a nitrogen stream. The Pt counter electrode was prepared as follows. First, two drops of 5 mM H2PtCl6 in ethanol were dropped onto the cleaned FTO glass substrate, then the FTO glass was dried and annealed at 400 °C for 15 min. The cell was fabricated by sealing the photoelectrode and Pt electrode together with a thermal plastics spacer with a diameter of 7 mm hole (Surlyn 1702, 25 μm, Solaronix) at 138 °C. The liquid electrolytes were injected into the space between the photoelectrode and Pt counter electrode through the hole on Pt counter electrode. The resultant cells were placed in a vacuum to remove air and to guarantee optimum filling and good electrical contact. These devices were sealed with a Surlyn sheet and thin glass by heating.
Characterization and photovoltaic measurements
1H NMR spectra were recorded on a Varian 400 MHz spectrometer. CHI660c electrochemical workstation was used to measure the cyclic voltammetry (CV). Three electrodes, working electrode-Pt disc, counter electrode-Pt wire, and reference electrode-Ag/Ag+. The Ag/Ag+ electrode was calibrated by ferrocene/ferrocenium (Fc/Fc+). Lithium perchlorate (LiClO4, 0.1 M) was used as the supporting electrolyte in acetonitrile. TU1800 SPC spectrometer was used to do the test of UV-vis spectra.
CHI660c electrochemical workstation was used to obtain the electrochemical impedance spectra (EIS) of the cells through using an AC impedance method at a forward bias voltage of −0.7 V for the impedance measurement over 0.01–105 Hz under dark conditions with an amplitude is 5 mV; resistances were calculated from the values between abscissa of three semi-circulars in the Nyquist plots. Mott–Schottky plots were obtained using a CHI660c electrochemical workstation at 10 Hz. The photocurrent density–voltage (J–V) curves of the assembled DSSCs shielded by an aluminum foil mask with an aperture area of 0.1 cm2 were measured with a digital light source intensity meter (Keithley, model 2612) under simulated air mass (AM) 1.5 solar spectrum conditions at 15, 50, and 100 mW cm−2, respectively. Incident photo-to-current conversion efficiency (IPCE) plotted as a function of excitation wavelength was recorded on a Keithley 2612 source meter under irradiation of a xenon lamp with a monochromater (Oriel Cornerstone™ 260 1/4).
Results and discussion
Redox potential and UV-vis spectra of redox couple
Scheme 1 shows the synthetic route and chemical structures of BMIT and BMIDT. The chemical structure and purity of the synthesized compounds were confirmed by 1H NMR (see Experimental section). It has already been confirmed that the redox potential plays a key role in determining the photovoltaic performance of DSSCs.29 In this work, cyclic voltammetry (CV) method using 0.05 M LiClO4 as the supporting electrolyte was applied to study the redox potentials of the BMIT/BMIDT. The obtained scans were calibrated with ferrocene/ferrocenium (Fc/Fc+), as shown in Fig. 1a. It can be clearly seen that BMII/I2 solution exhibits a more negative reduction peak (marked with ‘*’) than that of BMIT/BMIDT due to the reduction of triiodide:
 |
| | Fig. 1 (a) Cyclic voltammogram of redox couples in acetonitrile solution containing 0.05 M LiClO4 as the supporting electrolyte. The solutions that were studied containing 2 mM BMII + 2 mM I2 or 2 mM BMIT + 2 mM BMIDT in acetonitrile solution carried out at 20 °C. Scan rate: 50 mV s−1. (b) Energy levels of DSSC components, approximate redox potentials and band energies of the different components. | |
The redox potential versus the normal hydrogen electrode (NHE) was calibrated by adding a constant of 630 mV.39 Here, the standard redox potential of BMIT/BMIDT was determined to be +0.72 V vs. NHE, which is about 0.37 V higher than that of I−/I3− redox couple (i.e., about +0.35 V vs. NHE). Since the redox potential of dye N719 and I−/I3− are about +1.12 V and +0.35 V versus NHE,6 respectively, an excessive driving force of 0.77 V for regeneration process is needed. The energy loss incurred during dye regeneration is one of the main factors which limits the performance of these DSSCs.6,31 For instance, 0.6 eV for dye regeneration was calculated for an N719-sensitized TiO2-based DSSC using a I−/I3− redox couple (a driving force of 0.15 eV for electron injection does not seem to be very high).40 Therefore, it was assumed that the BMIT/BMIDT redox couple has a higher redox potential for DSSCs, which will lead to a higher Voc of the cells than that of the I−/I3− redox couple.41
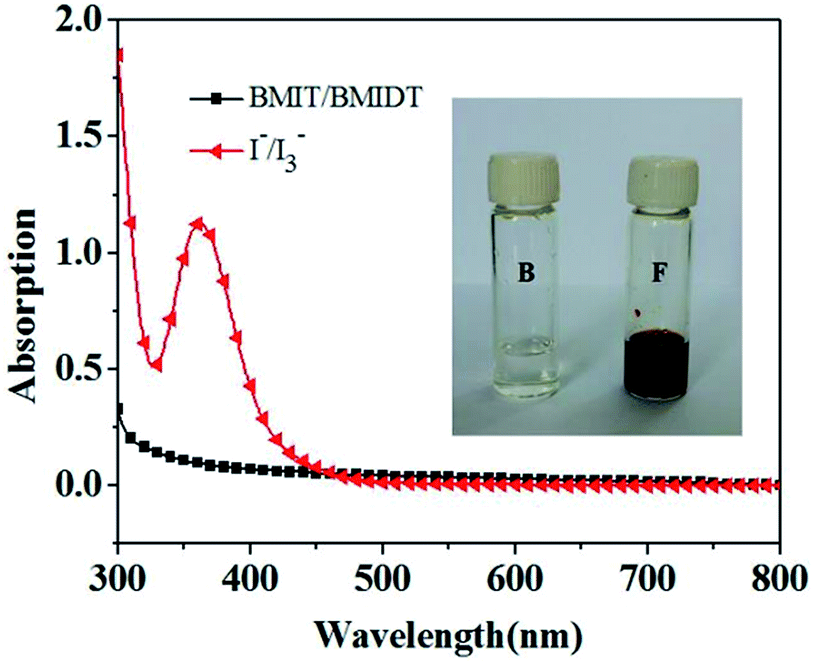
Fig. 2 shows the UV-vis spectra of BMIT/BMIDT and I−/I3− redox couples, respectively. As can be seen the I−/I3− redox couple exhibits strong absorption up to 500 nm owing to the presence of I3− in the electrolyte, while the BMIT/BMIDT shows a quite weak absorption in the same region. The weak absorption in the visible region of BMIT/BMIDT decreases the light absorption competition with the sensitizer, which is of great benefit to the utilization of solar energy. The inset of Fig. 2 shows the photograph of the electrolytes B and F. The colour of BMII/I2 based electrolyte F is much deeper than that of the iodine-free based electrolyte B, which is consistent with the UV-vis spectra.
 |
| | Fig. 2 UV-vis spectra of BMIT/BMIDT (0.1 mM/0.1 mM, represents 1000 times diluted electrolyte B); and BMII/I2 (0.1 mM/0.1 mM, represents 1000 times diluted electrolyte F) in acetonitrile solutions. Inset shows the photo of electrolytes B and F. | |
Photovoltaic performance
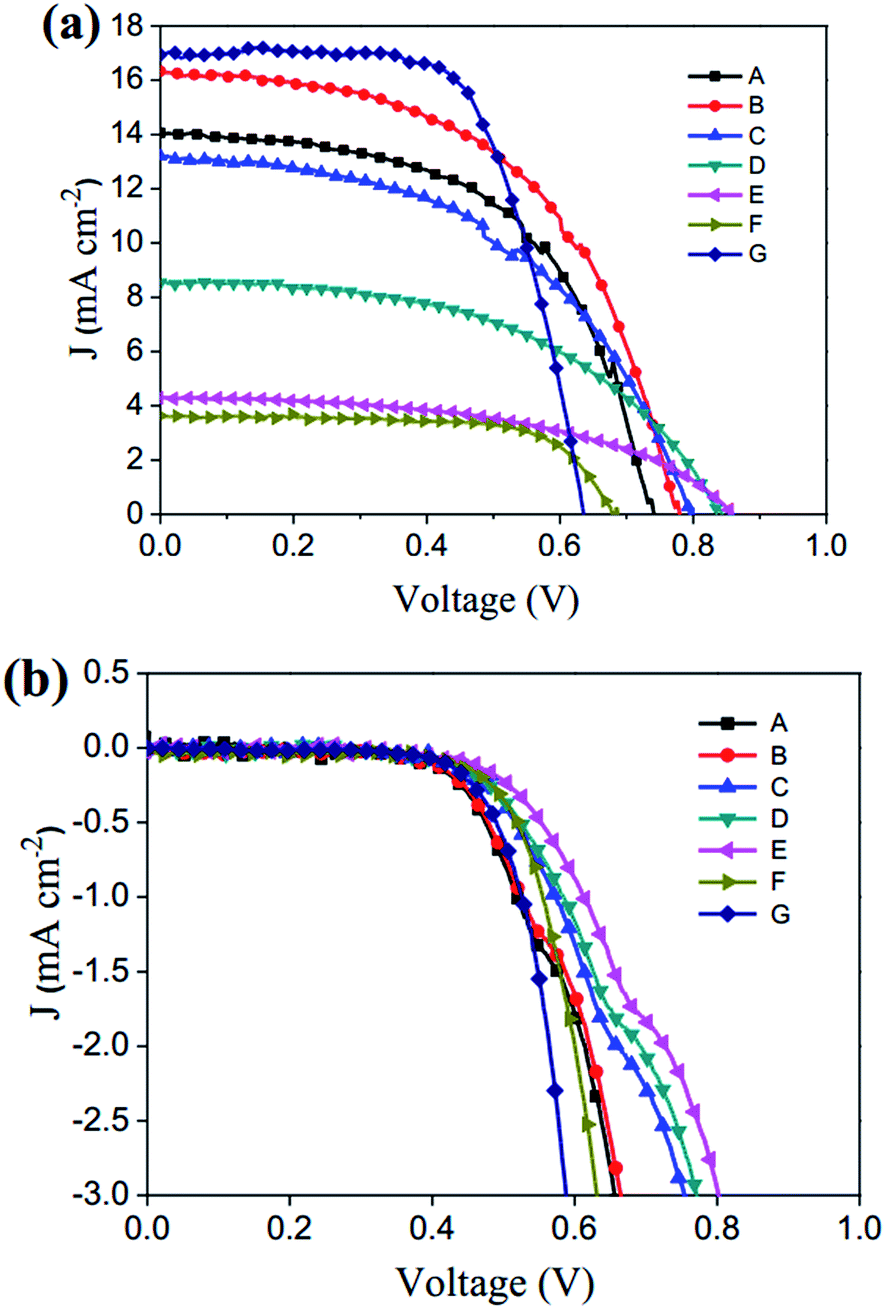
Fig. 3 shows the photocurrent density–voltage (J–V) curves of the fabricated DSSCs containing electrolytes A–F (cells A–G) under simulated AM 1.5 solar spectrum illumination of 100 mW cm−2 as well as in the dark. The relative parameters Voc, Jsc, FF, and PCE of the DSSCs are listed in Table 2. It can be clearly seen that the cells A–E show higher Jsc values than does cell F with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of BMII and I2. These results may be due to the strong visible light absorption of electrolyte F which decreases the light harvesting of the photosensitizer. Moreover, the cells A–E show distinctly higher Voc if compared with cells F and G due to the higher redox potential of BMIT/BMIDT, which reduces the energy loss incurred during dye regeneration. The relative CV curves of cells A–E were also studied in Fig. S2.† All the cells almost show the same redox potential, further suggest that cells based on BMIT/BMIDT have a higher Voc.
:1 molar ratio of BMII and I2. These results may be due to the strong visible light absorption of electrolyte F which decreases the light harvesting of the photosensitizer. Moreover, the cells A–E show distinctly higher Voc if compared with cells F and G due to the higher redox potential of BMIT/BMIDT, which reduces the energy loss incurred during dye regeneration. The relative CV curves of cells A–E were also studied in Fig. S2.† All the cells almost show the same redox potential, further suggest that cells based on BMIT/BMIDT have a higher Voc.
 |
| | Fig. 3 The J–V curves of DSSCs containing electrolytes A–F at 20 °C, (a) under simulated AM 1.5 solar spectrum irradiation at 100 mW cm−2, and (b) in dark. Cells are tested using an aluminum foil mask with an aperture area of 0.1 cm2. | |
Table 2 Photovoltaic performance parameters of DSSCs with different electrolytes under simulated AM 1.5 solar spectrum illumination at 100 mM cm−2 (average of four cells)
| Electrolyte |
Jsc (mA cm−2) |
Voc (V) |
FF |
PCE (%) |
| A |
14.06 (±0.13) |
0.74 (±0.01) |
0.56 (±0.01) |
5.82 (±0.16) |
| B |
16.11 (±0.22) |
0.77 (±0.01) |
0.54 (±0.02) |
6.70 (±0.28) |
| C |
13.20 (±0.29) |
0.80 (±0.02) |
0.51 (±0.02) |
5.39 (±0.21) |
| D |
8.54 (±0.16) |
0.84 (±0.01) |
0.51 (±0.03) |
3.64 (±0.34) |
| E |
4.25 (±0.24) |
0.86 (±0.03) |
0.50 (±0.02) |
1.83 (±0.24) |
| F |
3.63 (±0.19) |
0.68 (±0.01) |
0.68 (±0.01) |
1.69 (±0.12) |
| G |
16.94 (±0.174) |
0.63 (±0.02) |
0.69 (±0.02) |
7.39 (±0.18) |
The effects of the BMIT/BMIDT ratio on the photovoltaic properties of these devices were further studied. It can be clearly seen that the Voc value increases with decreasing BMIT/BMIDT ratio (cells A–D), because of the suppression of the dark current at the TiO2/electrolyte interface, as shown in Fig. 3b.26 It should be noted that cell B (BMIT/BMIDT, 0.1 M/0.1 M), based on an equal molar ratio of BMIT/BMIDT exhibited a Jsc of 16.11 mA cm−2, Voc of 770 mV, FF of 0.54, and yielded the highest photoelectric conversion efficiency of 6.70% under simulated AM 1.5 solar spectrum illumination at 100 mW cm−2. To the best of our knowledge, this is the lowest concentration of iodine-free redox couple reported that achieves a comparable PCE.
The effect of BMIT/BMIDT concentration ratios on performance was studied further (see ESI, Table S1 and Fig. S3†). Compared with cell B, the cells based on the relatively higher (0.4 M/0.4 M and 0.2 M/0.2 M) or lower (0.05 M/0.05 M) concentration ratios of BMIT/BMIDT (cells G–I) exhibited lower Jsc and PCE values, especially for cell G. It should be noted that the high concentration of BMIT/BMIDT (0.4 M/0.4 M) led to an obvious desorption of photosensitizer from the TiO2 photoelectrode, as shown in Fig. 4. Similar results were reported in the literature.42 The dye colour of the photoelectrode of cell G (BMIT/BMIDT, 0.4 M/0.4 M) is lighter than that of cell B (BMIT/BMIDT, 0.1 M/0.1 M), due to the desorption of photosensitizer in cell G. It has already been demonstrated that dye N719 could be adsorbed on the TiO2 surface through the carboxyl groups.43–45 Therefore, it is not surprising that both BMIT and BMIDT could adsorb the TiO2 photoelectrode surface through the carboxyl groups. The high concentration BMIT/BMIDT redox couple (for example at 0.4 M/0.4 M) will desorb the dye N719 from the TiO2 surface, and thus decrease the PCE values in cell G. Here, the electrolyte B can be considered as optimized with respect to the BMIT/BMIDT redox couple that exhibits the best efficiency among all the non-iodine based electrolytes studied in this work.
 |
| | Fig. 4 The photo of well-sealed cells containing cell B (BMIT/BMIDT, 0.1 M/0.1 M) and cell G (BMIT/BMIDT, 0.4 M/0.4 M). | |
The electron transfer process is hypothesized to occur as follows: under illumination, the optically excited dye (eqn (1)) injects an electron into the conduction band of TiO2 (eqn (2)) and transfer to the circuit load, and then return to the cathode. Then the oxidized dye is reduced to its ground by the electron transfer process is hypothesized to occur as follows: under illumination, the optically excited dye (eqn (1)) injects an electron into the conduction band of TiO2 (eqn (2)) and transfer to the circuit load, and then return to the cathode. Then the oxidized dye is reduced to its ground by T− (where T− is the thiolate anion) forming a radical cation (T˙) (eqn (3)). When two T˙ radical cations are formed, they combine to form T22+ (eqn (4)). Finally, the T2 gets reduced by electrons at cathode (eqn (5)). This possible redox scenario is shown as following reactions:35
| | |
dye* → dye+ + e− (CB)
| (2) |
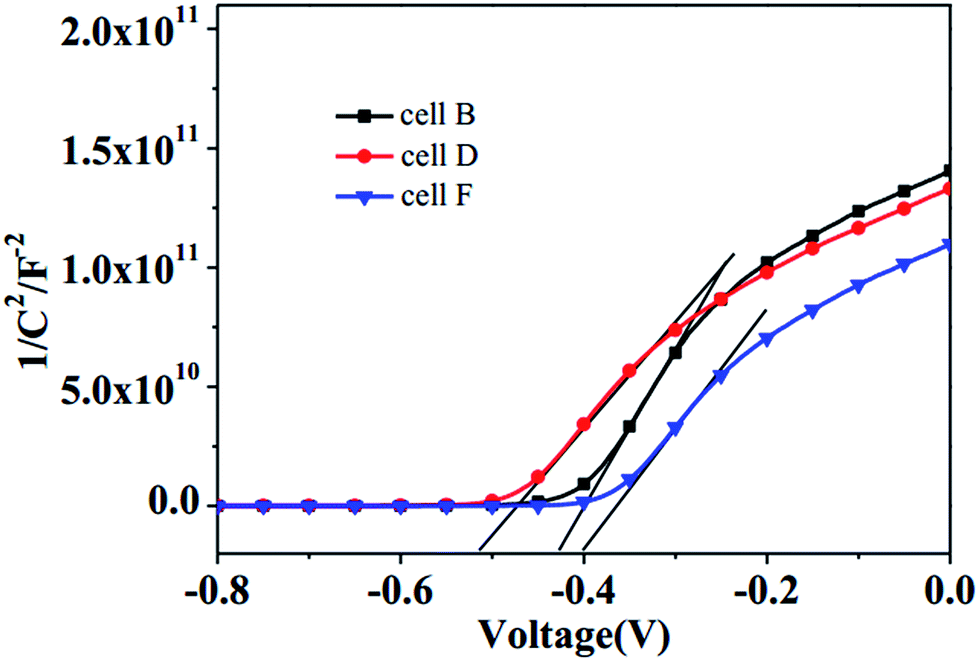
In order to further explain the Voc values of BMIT/BMIDT redox couples, Mott–Schottky plots of the TiO2 electrodes with electrolyte B, D, F were made to determine the shift of the conduction band (CB) edge of the TiO2 photoelectrode,46,47 as shown in Fig. 5. The Voc was derived from the equation Voc = |EF,n − Eredox|, where EF,n is the quasi-Fermi level and Eredox is the redox potential of the electrolyte. The negative shift in potential of the CB edge of TiO2 means a shift away from the BMIT/BMIDT redox potential, and an increase of Voc. The potentials of the CB edge of TiO2 with electrolytes B and D are negatively shifted about 0.03 V and 0.11 V, respectively, more than that of the iodine-based electrolyte F. Therefore, the theoretical voltages for using these electrolytes should decrease in the order D > B > F. These results appear consistent with the photovoltaic parameters of the devices.
 |
| | Fig. 5 The Mott–Schottky of TiO2 with electrolytes B, D, and F. | |
Fig. 6 shows the incident photon-to-current conversion efficiency (IPCE) values of devices based on the electrolytes B, D, and F. The maximum IPCE values at around 530 nm are 82.0%, 63.8% and 65.3% for the cells with the electrolytes B, D, and F, respectively. However, between 350 and 450 nm, the IPCE values of electrolytes B and D are higher than that of electrolyte F, most likely due to the strong light absorption of electrolyte F in this part of the visible spectrum. Moreover, the IPCE behavior in this region is consistent with the variation of Jsc. Similar results were observed with electrolytes G, H, I (Fig. S4†). The PCE values of devices prepared with electrolytes B, D, and F under different illumination intensities are summarized in Table 3. It can be noted that the PCE values of cells at 0.5 sunlight is higher than that at 1.0 sunlight, because of the inefficient charge screening of electron transport within the TiO2 film,48 while the PCE at 0.15 sunlight is lower than that at 0.5 sunlight due to the photocurrent response to light intensities and the changes in performance parameters, such as a lowered Voc and FF.49 The DSSC based on electrolyte B shows a maximum PCE value of 8.10% under AM 1.5 solar spectrum illuminations at 50 mW cm−2.
 |
| | Fig. 6 The IPCE vs. wavelength profiles for DSCCs based electrolytes B, D and F. | |
Table 3 The PCE of DSSCs based on different electrolytes under simulated AM 1.5 solar irradiation (average of four cells)
| Device |
PCE under different incident light intensities irradiation |
| 1.0 sun |
0.5 sun |
0.15 sun |
| Cell B |
6.78% (±0.28) |
8.10% (±0.17) |
8.00% (±0.22) |
| Cell D |
3.64% (±0.34) |
5.49% (±0.23) |
4.96% (±0.28) |
| Cell F |
1.69% (±0.12) |
2.14% (±0.11) |
1.93% (±0.12) |
Electrochemical impedance spectra
Electrochemical impedance spectroscopy (EIS) was used to investigate the interfacial charge transfer processes in DSSCs.50 The cells based on electrolytes B, D, and F were measured at −0.7 V bias in a dark environment, as shown in Fig. 7. The high frequency semicircle and medium frequency semicircle represent the charge-transfer resistance at the counter electrode (RCE) and at the TiO2/dye/electrolyte interface (Rrec), respectively.51 The values obtained for RCE and Rrec are summarized in Table 4. Fig. 7b shows Bode phase plots of these EIS spectra which display the characteristic frequency peaks of the charge transfer process for each of the cells. The effective lifetime of electrons (τe) before recombination in the TiO2 photoelectrode can be related to the inverse of the characteristic frequency and can be estimated using the following equation:52,53| |
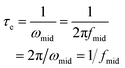 | (6) |
where ωmid is the angular frequency of the middle-frequency peak, and fmid is the frequency of the middle-frequency peak in Hz. The fmid and lifetime of electrons are also summarized in Table 4. We can see that the RCE values of cells B and D are higher than that of cell F, indicating that the oxidized species of this organic redox couple is regenerated slowly on the surface of the Pt counter electrode when compared to I−/I3− redox couple, which results in a lower FF.54 Therefore, more study is needed to seek a suitable catalyst to improve the fill factor of cells using this type of redox couples.55 The Rrec and τe of cell B are 599.50 Ω and 95.30 ms, respectively. The corresponding values for the cell F are much lower, amounting to 127.52 Ω and 7.39 ms, respectively, indicating that the recombination reaction between the conduction band electrons in TiO2 photoanode and BMIT/BMIDT-based electrolyte B is better suppressed than that with the iodine-based electrolyte F.31 Compared with cell F, the distinctly lower recombination rate between TiO2 and electrolyte B is partially responsible for the higher Voc of cell B, as well as for cell D (Table 4). Furthermore, the Rrec and τe values of cells G, H, and I were calculated to be 691.79, 364.52, 305.27 Ω, and 131.10, 96.53, 58.42 ms, respectively (Fig. S5†). These values are higher than the corresponding values for the cell F, which further demonstrate that the electron recombination reaction at the surface of TiO2 film and BMIT/BMIDT based electrolyte is more difficult than that of iodine-based electrolyte.
 |
| | Fig. 7 (a) Nyquist plots of the electrochemical impedance spectra measured for DSSCs based on electrolytes B, D, and F; (b) fitted Bode phase plots of each device. The impedance measurement was conducted at the bias voltage of −0.70 V and the frequency ranged from 0.01–105 Hz in dark. | |
Table 4 Parameters obtained by fitting the EIS of the DSSCs fabricated with different electrolytes
| Electrolyte |
RCE/Ω |
Rrec/Ω |
fmid/Hz |
τe/ms |
| B |
28.86 |
599.50 |
1.67 |
95.30 |
| D |
47.79 |
967.72 |
1.19 |
133.74 |
| F |
7.36 |
127.52 |
21.55 |
7.39 |
Long-term stability
The stability of these DSSCs was investigated under dark conditions at RT. The total efficiencies are normalized to the values measured on the first day, as shown in Fig. 8. The cell F containing I−/I3− based electrolyte still retained almost 50% of its initial conversion efficiency after testing for 1008 h. However, the cells with BMIT/BMIDT based electrolyte suffered a large decrease in PCE. For example, cell B retained about 27% of its initial conversion efficiency after 1008 h test. This behaviour may be attributed to the deactivation of the electroactive platinum layer on the counter electrode with time.56 Another possible reason is slight desorption of photosensitizer caused by electrolyte. Therefore, a more efficient electrocatalyst for the reduction of thiolate/disulfide redox couple at the counter electrode will be needed to extend the service life of such DSSCs.
 |
| | Fig. 8 The stability tests of DSSCs containing electrolytes B, D, and F during 1008 h storage at dark conditions and RT. | |
Conclusions
Organic thiolate/disulfide redox couples based on thiopropionic acid were used in combination with dye N719 in DSSCs to obtain a lower absorption of visible light and higher redox potential, relative to using I−/I3− redox couples. In addition, this organic redox couple resulted in a higher circuit current density and Voc. The resultant DSSCs containing an optimized ratio of BMIT/BMIDT (0.1 M/0.1 M) in combination with dye N719 exhibited a high overall PCE of 8.10% under AM 1.5 solar illumination at 50 mW cm−2. However, this BMIT/BMIDT based electrolyte requires a more efficient electrocatalyst for its regeneration.
Acknowledgements
This work was supported by the National Science Foundation for Distinguished Young Scholars (No. 21425417), the Natural Science Foundation of China (No. 21274101), the National Basic Research Program of China (973 Program) (No. 2012CB825800), and the Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Notes and references
- B. O'regan and M. Grfitzeli, Nature, 1991, 353, 737–740 CrossRef.
- T. Deepak, G. Anjusree, S. Thomas, T. Arun, S. V. Nair and A. S. Nair, RSC Adv., 2014, 4, 17615–17638 RSC.
- W. Wang, X. Guo and Y. Yang, Electrochim. Acta, 2011, 56, 7347–7351 CrossRef CAS.
- K. Ladomenou, T. Kitsopoulos, G. Sharma and A. Coutsolelos, RSC Adv., 2014, 4, 21379–21404 RSC.
- J.-H. Yum, E. Baranoff, F. Kessler, T. Moehl, S. Ahmad, T. Bessho, A. Marchioro, E. Ghadiri, J.-E. Moser and C. Yi, Nat. Commun., 2012, 3, 631 CrossRef PubMed.
- T. Daeneke, T.-H. Kwon, A. B. Holmes, N. W. Duffy, U. Bach and L. Spiccia, Nat. Chem., 2011, 3, 211–215 CrossRef CAS PubMed.
- Y. Bai, Q. Yu, N. Cai, Y. Wang, M. Zhang and P. Wang, Chem. Commun., 2011, 47, 4376–4378 RSC.
- J. Fan, Y. Hao, A. Cabot, E. M. Johansson, G. Boschloo and A. Hagfeldt, ACS Appl. Mater. Interfaces, 2013, 5, 1902–1906 CAS.
- D. Xu, H. Zhang, X. Chen and F. Yan, J. Mater. Chem. A, 2013, 1, 11933–11941 CAS.
- S. Y. Brauchli, F. J. Malzner, E. C. Constable and C. E. Housecroft, RSC Adv., 2014, 4, 62728–62736 RSC.
- J. Cong, Y. Hao, G. Boschloo and L. Kloo, ChemSusChem, 2015, 8, 264–268 CrossRef CAS PubMed.
- H. J. Snaith, S. M. Zakeeruddin, Q. Wang, P. Pechy and M. Graetzel, Nano Lett., 2006, 6, 2000–2003 CrossRef CAS PubMed.
- A. Sepehrifard, B. A. Kamino, T. P. Bender and S. Morin, ACS Appl. Mater. Interfaces, 2012, 4, 6211–6215 CAS.
- J. Xia, N. Masaki, M. Lira-Cantu, Y. Kim, K. Jiang and S. Yanagida, J. Am. Chem. Soc., 2008, 130, 1258–1263 CrossRef CAS PubMed.
- E. Premalal, N. Dematage, G. Kumara, R. Rajapakse, M. Shimomura, K. Murakami and A. Konno, J. Power Sources, 2012, 203, 288–296 CrossRef CAS.
- L. L. Sun, T. Zhang, J. Wang, H. Li, L. K. Yan and Z. M. Su, RSC Adv., 2015, 5, 39821–39827 RSC.
- Z. Huang, X. Zeng, H. Wang, W. Zhang, Y. Li, M. Wang, Y.-B. Cheng and W. Chen, RSC Adv., 2014, 4, 60670–60674 RSC.
- I. R. Perera, T. Daeneke, S. Makuta, Z. Yu, Y. Tachibana, A. Mishra, P. Bäuerle, C. A. Ohlin, U. Bach and L. Spiccia, Angew. Chem., Int. Ed., 2015, 54, 3758–3762 CrossRef CAS PubMed.
- D. Ameline, S. Diring, Y. Farre, Y. Pellegrin, G. Naponiello, E. Blart, B. Charrier, D. Dini, D. Jacquemin and F. Odobel, RSC Adv., 2015, 5, 85530–85539 RSC.
- C. Teng, X. Yang, C. Yuan, C. Li, R. Chen, H. Tian, S. Li, A. Hagfeldt and L. Sun, Org. Lett., 2009, 11, 5542–5545 CrossRef CAS PubMed.
- X. Wu, J. Zheng and C. Xu, Electrochim. Acta, 2016, 191, 902–907 CrossRef CAS.
- G. Oskam, B. V. Bergeron, G. J. Meyer and P. C. Searson, J. Phys. Chem. B, 2001, 105, 6867–6873 CrossRef CAS.
- B. V. Bergeron, A. Marton, G. Oskam and G. J. Meyer, J. Phys. Chem. B, 2005, 109, 937–943 CrossRef CAS PubMed.
- M. Cheng, X. Yang, F. Zhang, J. Zhao and L. Sun, Angew. Chem., Int. Ed., 2012, 124, 10034–10037 CrossRef.
- Z. Yu, C. Bu, Z. Zhou, Y. Liu, N. Huang, S. Bai, H. Fu, S. Guo and X. Zhao, Electrochim. Acta, 2013, 107, 695–700 CrossRef CAS.
- Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2008, 18, 341–346 CrossRef CAS.
- J. Min, J. Won, Y. S. Kang and S. Nagase, J. Photochem. Photobiol., A, 2011, 219, 148–153 CrossRef CAS.
- F. Kato, N. Hayashi, T. Murakami, C. Okumura, K. Oyaizu and H. Nishide, Chem. Lett., 2010, 39, 464–465 CrossRef CAS.
- W. Zhang, L. Qiu, X. Chen and F. Yan, Electrochim. Acta, 2014, 117, 48–54 CrossRef CAS.
- J. Cong, X. Yang, L. Kloo and L. Sun, Energy Environ. Sci., 2012, 5, 9180–9194 CAS.
- M. Cheng, X. Yang, S. Li, X. Wang and L. Sun, Energy Environ. Sci., 2012, 5, 6290–6293 CAS.
- S. Powar, R. Bhargava, T. Daeneke, G. Götz, P. Bäuerle, T. Geiger, S. Kuster, F. A. Nüesch, L. Spiccia and U. Bach, Electrochim. Acta, 2015, 182, 458–463 CrossRef CAS.
- H. Tian, Z. Yu, A. Hagfeldt, L. Kloo and L. Sun, J. Am. Chem. Soc., 2011, 133, 9413–9422 CrossRef CAS PubMed.
- H. Tian and L. Sun, J. Mater. Chem., 2011, 21, 10592–10601 RSC.
- M. Wang, N. Chamberland, L. Breau, J.-E. Moser, R. Humphry-Baker, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Nat. Chem., 2010, 2, 385–389 CrossRef CAS PubMed.
- H. Tian, X. Jiang, Z. Yu, L. Kloo, A. Hagfeldt and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 7328–7331 CrossRef CAS PubMed.
- B. Qiu, C. Pan, W. Qian, Y. Peng, L. Qiu and F. Yan, J. Mater. Chem. A, 2013, 1, 6373–6378 CAS.
- Y. Zhang, Z. Sun, S. Cheng and F. Yan, ChemSusChem, 2016, 9, 813–819 CrossRef CAS PubMed.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef CAS.
- T. Stergiopoulos and P. Falaras, Adv. Energy Mater., 2012, 2, 616–627 CrossRef CAS.
- M. Wang, N. Chamberland, L. Breau, J.-E. Moser, R. Humphry-Baker, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Nat. Chem., 2010, 2, 385–389 CrossRef CAS PubMed.
- H. Tian, E. Gabrielsson, P. W. Lohse, N. Vlachopoulos, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2012, 5, 9752–9755 CAS.
- K. E. Lee, M. A. Gomez, S. Elouatik and G. P. Demopoulos, Langmuir, 2010, 26, 9575–9583 CrossRef CAS PubMed.
- C. Pérez León, L. Kador, B. Peng and M. Thelakkat, J. Phys. Chem. B, 2006, 110, 8723–8730 CrossRef PubMed.
- A. Byrne, N. J. English, U. Schwingenschlögl and D. F. Coker, J. Phys. Chem. C, 2015, 120, 21–30 Search PubMed.
- M.-H. Kim and Y.-U. Kwon, J. Phys. Chem. C, 2009, 113, 17176–17182 CAS.
- L. Li, Y. Hao, X. Yang, J. Zhao, H. Tian, C. Teng, A. Hagfeldt and L. Sun, ChemSusChem, 2011, 4, 609–612 CrossRef CAS PubMed.
- Z. Sun, M. Liang and J. Chen, Acc. Chem. Res., 2015, 48, 1541–1550 CrossRef CAS PubMed.
- C. Shi, L. Qiu, X. Chen, H. Zhang, L. Wang and F. Yan, ACS Appl. Mater. Interfaces, 2013, 5, 1453–1459 CAS.
- X. Chen, Q. Li, J. Zhao, L. Qiu, Y. Zhang, B. Sun and F. Yan, J. Power Sources, 2012, 207, 216–221 CrossRef CAS.
- S. Li, L. Qiu, C. Shi, X. Chen and F. Yan, Adv. Mater., 2014, 26, 1266–1271 CrossRef CAS PubMed.
- R. Kern, R. Sastrawan, J. Ferber, R. Stangl and J. Luther, Electrochim. Acta, 2002, 47, 4213–4225 CrossRef CAS.
- H. Li, S. Li, Y. Zhang and F. Yan, RSC Adv., 2016, 6, 346–352 RSC.
- J. Liu, X. Yang, J. Cong, L. Kloo and L. Sun, Phys. Chem. Chem. Phys., 2012, 14, 11592–11595 RSC.
- H. Wu, Z. Lv, S. Hou, X. Cai, D. Wang, H. Kafafy, Y. Fu, C. Zhang, Z. Chu and D. Zou, J. Power Sources, 2013, 221, 328–333 CrossRef CAS.
- J. Burschka, V. Brault, S. Ahmad, L. Breau, M. K. Nazeeruddin, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Energy Environ. Sci., 2012, 5, 6089–6097 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of devices. See DOI: 10.1039/c6ra11592f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.