
Ab initio solute–interstitial impurity interactions in vanadium alloys: the roles of vacancy
Abstract
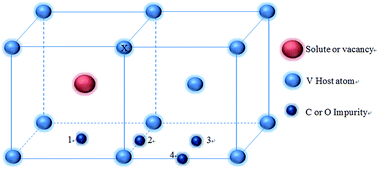
This study aims to characterize the interactions between substitutional solutes (3d, 4d and 5d transition metals) and interstitial impurities (C and O) in vanadium alloys, with or without the presence of an adjacent vacancy. For this purpose, the binding energies for solute–impurity and vacancy–impurity pairs, as well as solute–vacancy–impurity complexes are investigated by means of first-principles calculations, with or without the elastic correction. The vacancy–impurity binding energies suggest that it is energetically favorable to form stable 1nn vacancy–impurity pairs. For large-sized solutes, the solute–impurity interactions present strong repulsive interactions when a vacancy is absent, while showing strong attractive ones in the presence of a vacancy. Furthermore, a comprehensive study on the binding energy of defects revealed a positive correlation between the elastic correction energies and solute volumes, indicating that the elastic correction for the binding energies needs to be considered when a vacancy is absent in the vicinity of defects. Based on the binding preference, we can infer that a vacancy prefers to bond with large solutes adjacent to it and thus the resulting solute–vacancy pair can serve as a strong impurity trapper to form a defect complex, enhancing the nucleation and growth of precipitates in V alloys.
 Please wait while we load your content...
Please wait while we load your content...

