
Three-dimensional graphene/Fe3O4-based magnetic solid phase extraction coupled with high performance liquid chromatography for determination of carvedilol in human blood plasma
Abstract
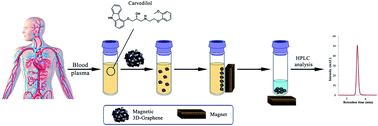
A magnetic three-dimensional graphene nano-composite (3D-G–Fe3O4) was synthesized and used as an effective adsorbent for the magnetic solid-phase extraction (MSPE) of carvedilol in human blood plasma samples followed by high performance liquid chromatography (HPLC) equipped with ultraviolet (UV) detection. The combination of magnetic nanoparticles with the porous structure of three dimensional graphene increases the adsorption capacity and surface area for extraction and determination of carvedilol in human blood plasma samples without the need of filtration or centrifugation. The properties and morphology of the synthesized adsorbent were characterized by Fourier transform-infrared spectroscopy (FT-IR), scanning electron microscopy (SEM), transmission electron microscope (TEM), vibrating sample magnetometry (VSM), Raman spectroscopy, X-ray diffraction (XRD), Brunauer–Emmett–Teller (BET), and Barrett–Joyner–Halenda (BJH) techniques. The main experimental parameters affecting the extraction recovery such as desorption conditions, pH of sample solution, dosage of adsorbent, extraction time, and salt concentration were investigated and optimized. Under the optimized experimental conditions, the figure of merit results showed an excellent linear dynamic range (2–1000 ng mL−1), with a determination coefficient (R2) higher than 0.994 and limits of detection (LOD) of 0.5 ng mL−1. Intra- and inter-day relative standard deviations (RSDs) were less than 2.70 and 3.85%, respectively.
 Please wait while we load your content...
Please wait while we load your content...

