DOI:
10.1039/C6RA11313C
(Paper)
RSC Adv., 2016,
6, 65572-65578
Molecules with O-acetyl group protect protein glycation by acetylating lysine residues†
Received
2nd May 2016
, Accepted 4th July 2016
First published on 4th July 2016
Abstract
Pharmaceutical intervention for reduction of advanced glycation end products (AGEs) is considered as a therapeutic strategy to attenuate the pathogenesis of diabetes. Many molecules have been reported to possess antiglycation activity, one such example is acetylsalicylic acid (aspirin). It protects proteins from glycation by acetylating the lysine residues. Therefore, in this study we have synthesized and screened molecules containing free N-acetyl, O-acetyl and acetophenone groups. All the selected molecules in this study showed glycation inhibition but interestingly, only molecules with O-acetyl but not N-acetyl and acetophenone groups were capable of acetylating lysine residue. Furthermore, we have demonstrated that pre-acetylation or aspirin treatment prior to the induction of diabetes helps in reducing HbA1c and AGE formation in the streptozotocin induced diabetic mice. Hence pre-acetylation may have an additional therapeutic efficacy of reducing AGE levels in vivo. Incorporation of O-acetyl group into anti-diabetic molecules could be a useful strategy, as it may have an additive effect in reducing AGEs. Identification of such novel acetylating agents represents a new area in the drug discovery process.
Introduction
Hyperglycemic condition in diabetes promotes formation of advanced glycation end products (AGEs), which are implicated in pathogenesis of diabetic complications including vascular stiffening,1 atherosclerosis,2 cataracts,3 and age associated disorders including Parkinson's disease,4 and Alzheimer's disease.5 Mounting evidence of involvement of AGEs in various diseases makes them a promising target for therapeutics. Hence, reducing AGE levels by both chemical and cellular approaches has been proposed as an intervention strategy to treat glycation associated diseases.6 Glycation can be intervened at various strategies viz. (i) Schiff's base formation, (ii) deglycation, (iii) transglycation, (iv) Amadori product inhibition, (v) crosslink inhibitors, (vi) crosslink breakers, (vii) RAGE (Receptor for AGEs) blockers etc. Apart from these strategies, formation of AGEs can also be prevented by blocking glycation sites, which can be achieved by modification of amino acid side chains resulting in e.g. acetylation, hydroxylation, methylation, formylation etc. Some of the preexisting FDA approved drugs possess anti glycation activity including aspirin, which is largely used for its anti-inflammatory, antipyretic, analgesic and anti-thrombotic effects. Aspirin in vitro readily acetylates crystallin, a lens protein, and protects it from glycation by galactose, glucosamine and glucose.7–9 In vivo studies with aspirin fed diabetic rats also showed decrease in crystallin glycation and HbA1c.9 Further, lysine acetylation contributes to protect the biological function of crystallin even in presence of glycation.10 In another in vivo study aspirin prevented inactivation of heme pathway enzymes δ-aminolevulinic dehydratase and porphobilinogen deaminase by glycation that would aid in preventing diabetic complications.11 Therefore, implication of lysine acetylation in various biological processes and complex association with cellular metabolism offers lot of clinical relevance. Recently, acetylation and glycation prone sites on HSA and other plasma proteins were characterized and quantified for better understanding of their interplay and reciprocal competition.12,13 Aspirin mediated acetylation and protein glycation influence each other through a dynamic equilibrium between acetylation and glycation.13 Therefore, acetylation plays a key role in regulation of protein glycation and molecules capable of acetylating could be useful in protection against glycation. Hence, in this study, we report the screening of acetyl group containing molecules for anti-glycation activity both in vitro and in vivo. Interestingly molecules having O-acetyl group (such as aspirin (ASP), bisacodyl (BIS) and NDS-458) are capable to acetylate the protein thereby decreasing the glycation than N-acetyl and acetophenone group. In addition, we have shown both in vitro and in vivo that pre-acetylation protects proteins from glycation.
Results and discussion
Screening and evaluation of acetyl group molecules for glycation inhibition
AGE-fluorescence assay. Molecules with acetyl group (Fig. 1) were evaluated for their anti-glycation activity by using BSA-AGE-fluorescence assay (excitation 370/emission 440 nm).14 The results indicate that all the molecules showed decrease in the relative fluorescence intensity in comparison with glycated BSA (Fig. 2A). However, aspirin and NDS-458 showed strong glycation inhibition (>60% with p < 0.05).
 |
| | Fig. 1 The molecular structures of acetyl group containing molecules. | |
 |
| | Fig. 2 (A) Anti-glycation activity by various molecules containing acetyl group as analyzed by BSA-AGE-fluorescence assay (excitation 370/emission 440 nm) (n = 3 ± SE). (B) Western blot analysis utilizing anti-AGE and anti-CML antibodies of (1) control BSA, (2) glycated BSA, (3) glycated BSA with ASP, (4) glycated BSA with BIS and (5) glycated BSA with NDS-458. | |
Western blot analysis. A qualitative and quantitative result of glycation inhibition by acetyl group containing molecules was also confirmed by western blot analysis utilizing anti-AGE and anti-CML antibodies. The analysis was performed to determine extent of BSA glycation both with and without incubation with acetyl group containing molecules. The results showed a clearly visible difference optical density of glycated BSA reflecting significant decrease in glycation extent upon incubation with ASP, BIS and NDS-458 (Fig. 2B and ESI Fig. 5†).
Insulin glycation assay. Insulin-MALDI based assay to screen the molecules for glycation inhibition was performed as described earlier.14 Insulin (5808 m/z) upon glycation with glucose (162 m/z) form glycated insulin (5970 m/z). The intensity of glycated insulin was monitored in presence of various acetyl group containing molecules [ASP, BIS, NDS-458, PAR, ACZ, ACH and NAC] to study the glycation inhibition activity. The results indicate that all the molecules showed decrease in the relative intensity of glycated insulin in comparison with control (Fig. 3A). ASP, BIS and NDS-458 showed strong glycation inhibition (>70% with p < 0.05).
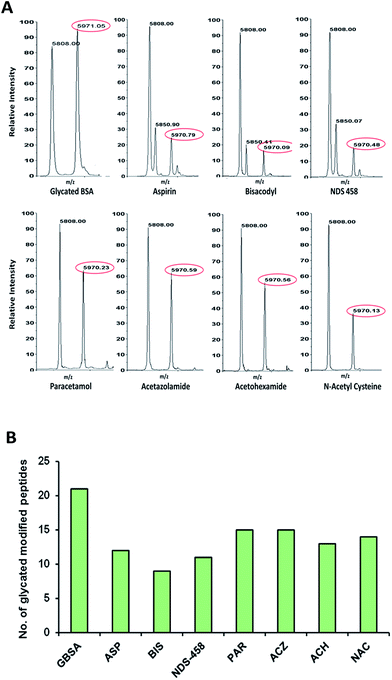 |
| | Fig. 3 (A) Insulin-MALDI based assay to screen the molecules for glycation inhibition; the relative intensity of glycated insulin was monitored in presence of various acetyl group containing molecules. (B) Bar graph depicting the number of glycated modified peptides of in vitro glycated BSA, glycated BSA in presence of various acetyl group containing molecules. | |
LC-MS/MS analysis of glycated BSA. Qualitative analysis of glycated BSA was complemented by quantitative LC-MS/MS analysis. The extent of BSA glycation inhibition by acetyl group containing molecules was measured as increase or decrease of glycation modified sites/peptides. This approach facilitated identification of the 21 Amadori modified peptides in glycated BSA. Whereas pre-glycated BSA incubated with acetyl group containing molecules showed decrease in the glycation. ASP, BIS, NDS-458, PAR, ACZ, ACH and NAC showed decrease in glycated peptides by 12, 9, 11, 15, 15, 13 and 14 respectively as shown in Fig. 3B, details of the modified peptides are listed in (ESI Table 1†). A representative MS/MS annotation of Amadori modified peptide of BSA is depicted in (ESI Fig. 3†). Taken together all the results (qualitative, quantitative and western blot) strongly support the glycation inhibition of acetyl group containing molecules.
Protein acetylation with acetyl group containing molecules
Protein acetylation assay. Insulin [m/z 5808] was used as a model protein to monitor the protein acetylation with acetyl group containing molecules. Formation of acetylated insulin [m/z 5850], which displays a characteristic mass shift of 42 Da corresponding to acetylation was monitored. Only three molecules ASP, BIS and NDS-458 were found to be acetylating the insulin as observed by formation of acetylated insulin peak, while remaining molecules could not cause acetylation of insulin (Fig. 4A). This was also reflected in BSA acetylation as analyzed by western blotting with anti-acetylation antibody (Fig. 4B and ESI Fig. 6†). The results showed clearly that, ASP, BIS and NDS-458 induced BSA lysine acetylation, amongst these three molecules, NDS-458 showed high intense band suggesting it strong acetylating capabilities (Fig. 4B).
 |
| | Fig. 4 (A) MALDI-TOF-MS based insulin acetylation inhibition assay with insulin in presence of various acetyl group containing molecules. (B) Western blot analysis utilizing anti-acetyl lysine antibody (1) BSA, (2) BSA with ASP, (3) BSA with BIS, (4) BSA with NDS-458, (5) BSA with paracetamol, (6) BSA with acetazolamide, (7) BSA with acetohexamide and (8) BSA with N-acetyl cysteine. | |
LC-MS/MS analysis of acetylated BSA. BSA acetylation was also studied by LC-MS/MS analysis. The site specific protein acetylation was monitored by using Q-Exactive MS. The extent of BSA acetylation by acetyl group containing molecules was measured as potential acetylating compound. ASP, BIS, and NDS-458 showed 43, 43, and 58 acetyl modified peptides of BSA respectively as listed in (ESI Table 2, ESI Fig. 7†). Whereas PAR, ACZ, ACH and NAC did not showed any acetyl modified BSA peptides. A representative MS/MS annotation of acetylated BSA peptide is shown in ESI Fig. 4.† NDS-458 molecule was synthesized by acetylating the hydroxyl group present in paracetamol. Taken together all the results (qualitative, quantitative and western blot) strongly support that O-acetyl group containing molecules (ASP, BIS, NDS-458) acetylates the lysine residues of protein rather than N-acetyl (PAR, ACZ, NAC) or acetophenone molecules (ACH).
Pre-acetylation protein assay. Pre-acetylated insulin and pre-acetylated BSA were used as model proteins to examine their ability to resist glycation. Pre-acetylated insulin showed a little glycation as assessed by MALDI-MS for the presence glycated peak (m/z 5970) corresponding to increase in mass shift 162 Da. However, incorporation of aspirin during glycation showed lesser resistance to glycation as there was formation of glycated insulin (Fig. 5A). Such results were also observed with pre-acetylated BSA, which showed more resistance to glycation as observed by lesser formation for glycated BSA products on SDS-PAGE (Fig. 5B). These results suggest that pre-acetylation is a better strategy in prevention of glycation.
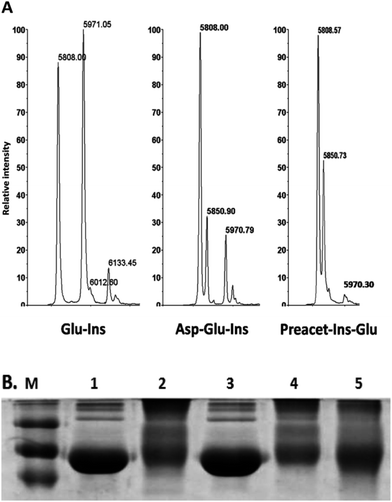 |
| | Fig. 5 (A) In vitro MALDI-TOF-MS based glycation assay in presence of pre-acetylation insulin with glucose. (B) SDS-PAGE analysis depicting resistance of pre-acetylated BSA to glycation of (1) control BSA, (2) glycated BSA, (3) aspirin treated BSA, (4) glucose treated with aspirin to BSA and (5) pre-aspirin treated BSA then glucose. | |
In vivo protective effect of pre-acetylation against glycation. Streptozotocin (STZ) induced diabetic mice models were utilized to demonstrate the protective effect of pre-acetylation against protein glycation. Pre-acetylation of proteins were attained by oral feeding of aspirin for a month prior to STZ injection. Mice models chosen for experiments were divided into five groups: group (i) healthy control; (ii) STZ induced diabetic; (iii) healthy control pre-treated with aspirin; (iv) STZ induced diabetic and aspirin treated and (v) aspirin pre-treated STZ induced diabetic. Acetylation was monitored by western-blotting with anti-acetylation antibody. Increased acetylation was observed in mice of both aspirin pre-treatment and post treatment of diabetes induction. This was accompanied by reduced AGE modified serum albumin as measured by western blotting with anti-CML antibody (Fig. 6A, ESI Fig. 8†). This result was also reflected in HbA1c levels (Fig. 6B).
 |
| | Fig. 6 (A) Western blot analysis utilizing anti-acetyl lysine and anti-CML antibodies in in vivo of plasma proteins (B) bar graph depicting the HbA1c (n = 6 ± SE) of (1) control mice, (2) diabetic mice, (3) aspirin treated control mice, (4) diabetic mice treated with aspirin and (5) pre-aspirin treated then STZ induced diabetic mice. | |
Experimental
Chemicals
All the chemicals were procured from Sigma-Aldrich (Sigma-Aldrich, MO, USA) unless otherwise are mentioned. Anti-AGE antibody, protein A-HRP conjugate were purchased from Merck Millipore (Merck Millipore, MA, USA). Anti-CML antibody was procured from Merck Abcam (Abcam, Cambridge, UK). Anti-acetylated lysine antibody was procured from Calbiochem (Calbiochem, CA, USA). MS grade water, acetonitrile (ACN), methanol (MeoH) and were procured from J. T. Baker (J. T. Baker, PA, USA).
Synthesis and characterization of 4-acetamidophenyl acetate (NDS-458)
To a solution of N-(4-hydroxyphenyl)acetamide (1.0 g, 6.6 mmol) and Et3N (1.1 mL, 7.9 mmol) in anhydrous CH2Cl2 (20 mL), acetyl chloride (0.62 mL, 7.9 mmol) was added drop wise at 0 °C and stirred for 2 h at the same temperature. The reaction mixture was diluted with CH2Cl2 (10 mL), washed with saturated NaHCO3 solution (5 mL), water (5 mL) and brine (5 mL). The organic layer was dried over NaSO4, concentrated under reduced pressure. The residue was purified by column chromatography (silica gel 100–200 mesh, MeOH–CH2Cl2 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95) to afford 4-acetamidophenylacetate (1.23 g, 97%) as a colorless solid. NDS-458 was characterized by 1H NMR and 13C NMR.
:95) to afford 4-acetamidophenylacetate (1.23 g, 97%) as a colorless solid. NDS-458 was characterized by 1H NMR and 13C NMR.
1H NMR (400 MHz, CDCl3) (ESI Fig. 1(S1)†): δ 7.71 (brs, 1H), 7.53–7.41 (m, J = 8.8 Hz, 2H), 7.09–6.91 (m, J = 9.0 Hz, 2H), 2.28 (s, 3H), 2.12 (s, 3H); 13C NMR (100 MHz, CDCl3) (ESI Fig. 2(S2)†): δ 169.9, 168.6, 146.7, 135.7, 121.9, 120.9, 24.4, 21.1. NMR values (1H & 13C) were compared with reported values and found to be identical.18
In vitro glycation of bovine serum albumin (BSA) and AGE fluorescence assay
The acetyl group containing molecules including aspirin (ASP), bisacodyl (BIS), NDS-458, paracetamol (PAR), acetazolamide (ACZ), N-acetyl cysteine (NAC) and acetohexamide (ACH) were selected for the screening of their in vitro glycation inhibition studies. 100 mM stock solutions of above mentioned molecules were prepared in 10% DMSO of 0.1 M phosphate buffer solution (PBS) (pH 7.4). 10% DMSO in 0.1 M PBS was used as a solvent control.
In vitro glycation of BSA was performed as described earlier.19 Briefly, 450 μl of BSA (50 mg mL−1) in 0.1 M phosphate buffer (pH 7.4) containing 225 μl of D-glucose (2 M), 5 mM sodium azide with or without 10 mM of all the above mentioned molecules were incubated at 37 °C for 15 days. The exact concentration of the bisacodyl in the final reaction was relatively lesser than other drugs (<10 mM) because of its solubility issue. BSA glycation was monitored by fluorescence spectroscopy, excitation at 370 and emission at 440 nm by using Varioskan Flash Multimode reader (Thermo Fisher Scientific, Germany).
Western blot analysis by using anti-AGE, anti-CML and anti-acetylated lysine antibodies
5 μg of control BSA, glycated BSA and glycated BSA incubated with 10 mM concentration acetyl group containing molecules [ASP, BIS, NDS-458, PAR, ACZ, ACH and NAC] for 15 days at 37 °C were resolved onto 12% SDS-PAGE and transferred onto the PVDF membrane. The membranes were blocked with 5% skimmed milk. The proteins were probed by anti-AGE, anti-CML and anti-acetylated lysine antibodies followed by incubation with protein A-HRP conjugate. Immunoreactive bands were visualized by using Western Bright ECL HRP substrate (Advansta, CA, USA) and documented by using gel doc (Syngene, Cambridge, UK).
Insulin glycation inhibition assay
The in vitro insulin glycation inhibition efficiency of acetyl group containing molecules [ASP, BIS, NDS-458, PAR, ACZ, ACH and NAC] was performed by using MALDI based insulin glycation inhibition assay. Briefly, 50 μl of insulin (1.8 mg mL−1) in 200 μl of 0.1 M phosphate buffer (pH 7.4) containing 50 μl of glucose (250 mM), 5 mM sodium azide with and without 10 mM concentration of all above mentioned acetyl group containing drugs were incubated for 7 days at 37 °C. The reaction mixture was mixed with sinapinic acid and analyzed on MALDI TOF/TOF 5800 (AB SCIEX, MA, USA) in linear mode as described.14
Insulin acetylation assay
Insulin (50 μl) (1.8 mg mL−1) was incubated with 10 mM concentration acetyl group containing molecules [ASP, BIS, NDS-458, PAR, ACZ, ACH and NAC] in 0.1 M phosphate buffer (pH 7.4) at 37 °C for 7 days. The reaction mixture was mixed with α-cyano-4-hydroxycinnamic acid and analyzed on MALDI TOF/TOF 5800 (AB SCIEX, MA, USA) in linear mode as described.20
Acetylation of BSA
The in vitro acetylation of BSA was performed by incubating 450 μl of BSA (50 mg mL−1) in 0.1 M phosphate buffer (pH 7.4) containing 5 mM sodium azide with or without 10 mM concentration acetyl group containing molecules [ASP, BIS, NDS-458, PAR, ACZ, ACH and NAC] at 37 °C for 3 days.14
Tryptic digestion
100 μg of protein was dissolved in 0.1% RapiGest and reduced with 100 mM dithiothreitol (DTT) at 60 °C for 15 min followed by alkylation with 200 mM iodoacetamide in dark at room temperature for 30 min. Then, proteins were subjected to tryptic digestion at 37 °C overnight. The digestion reaction was stopped by adding concentrated HCl and incubated for 10 min at 37 °C before vertex and centrifugation and stored at −80 °C till further use.
LC-HR/AM Q-Exactive Orbitrap analysis (full MS/dd-MS2)
Chromatographic separation. Peptides digest (1.5 μg) was separated on Hypersil Gold C18-RP HPLC column (150 × 2.1 mm, 1.9 μm) with 98% of mobile phase A (100% water, 0.1% FA) and 2% of mobile phase B (100% ACN, 0.1% FA) at 350 μl min−1 flow rate with a 45 min linear gradient of 2% to 40% mobile phase B.
MS acquisition. All samples were analyzed on Q-Exactive Orbitrap MS (Thermo Fisher Scientific, Germany). The instrument was tuned and calibrated for better performance. The tune parameters include: spray voltage 4200 V, capillary temperature 320 °C, heater temperature 200 °C, S-lens RF value 55, sheath and auxiliary gases pressure were 30 and 8 psi respectively. The samples were acquired in positive ionization (HESI) mode in data-dependent manner using a top-5 method with scan range from 350 to 1800 m/z. MS spectra were acquired at a resolution of 70000 with maximum injection time (IT) of 120 ms and automatic gain control (AGC) value of 1 × 106 ions and MS/MS spectra were acquired at 17500 resolution with maximum IT of 120 ms and AGC value of 1 × 105 ions. Precursor's selectivity was performed at isolation width of 2 m/z, under fill ratio of 0.3% and dynamic exclusion time of 15 s. The peptide fragmentation was performed using normalized high energy collision induced dissociation (30 eV).21
Database search and PTM analysis
The MS data set was processed using Proteome discoverer 1.4 (Version 1.4.0.288) (Thermo Fisher Scientific, Germany). SEQUEST HT search engine was used for peptide identification. The data was searched against UniProt BSA (P02769) sequence database. Carbamidomethylation of cysteine (C) and oxidation of methionine (M) was considered as fixed and variable modification respectively. Additional glycation modifications at lysine position Amadori (+162.05 Da), carboxymethyllysine (CML) (+58.005 Da), carboxyethyllysine (CEL) (+72.021 Da) and acetylation modification (+42.02 Da) were considered as variable modifications. The search was performed using the following parameters; peptide and fragment mass tolerance 10 ppm and 0.5 Da respectively, missed cleavages-2 and false discovery rate 1%. The glycated peptides were identified as described earlier.19,22
Animal experiments
The animal experiments were approved by Institutional Animal Ethics committee of National Centre for Cell Sciences, India. The experimental protocols were carried out in accordance with the guidelines of CPCSEA (Committee for the Purpose of Control and Supervision of Experiments on Animals), India. Male BALB/c mice were injected with 50 mg kg−1 body weight of streptozotocin (STZ) in 50 mM citrate buffer pH 4.5 for five consecutive days to induce hyperglycemia, whereas control mice were injected with 50 mM citrate buffer. The induction of diabetes was confirmed after 30 days by measuring the blood glucose levels with glucometer (Bayer, Germany) and HbA1c level by using HbA1c kit (Bayer, Germany). For each treatment six animals were grouped into control, diabetic, aspirin treated control, aspirin pre-treated diabetic and diabetic treated with aspirin (200 mg L−1) for 30 days after induction of diabetes. Aspirin was made available through drinking water. Glucose and HbA1c levels were monitored on 30th, 45th, and 60th day (0th, 15th and 30th day after initiation of STZ injection). Animals were euthanized at the end of study after 30 days of drug administration. Blood samples were collected; blood glucose and HbA1c were analyzed immediately. Plasma was obtained by EDTA treatment, which was then centrifuged at 1500g for 5 min, and the supernatant was stored at −80 °C until further use.23 Protein concentration was determined by using BioRad protein assay kit (BioRad, CA, USA).
Statistical analysis
All experiments were performed in triplicates. Data are expressed as mean ± SD. A p-value <0.05 was considered statistically significant.
Conclusions
Aspirin is being used in the treatment of cardiovascular diseases due to its antithrombotic effects through platelet-independent mechanisms.15 Previous studies have shown that aspirin has the ability to acetylate proteins.16 Therefore, in this study we investigated the protective effect of acetylation against glycation by synthesizing acetylating molecules and screened for their ability to protect glycation. Our results suggest that molecules with only O-acetyl but not N-acetyl group or acetophenone compounds were capable of acetylating lysine residue and protecting against glycation. The plausible mechanism for this observation could be amine reacts with O-acetyl carbonyl and then, O to N-acetyl transfer takes place with loss of corresponding phenolic derivative (ESI Fig. 9†). However, in the case of N-acetyl compounds (e.g. paracetamol) and acetophenone related compounds (acetohexamide), corresponding carbonyl groups are not reactive enough towards attack by the amines under normal physiological conditions, hence there is no acetyl migration was observed.24–26 Therefore, we propose that incorporation of O-acetyl group into anti-diabetic molecules could be a useful strategy, as it may have an additive effect in reducing AGEs. Identification of such novel acetylating agents represents a new area in the drug discovery process. Furthermore, pre-acetylation or aspirin treatment prior to the induction of diabetes helps in reducing HbA1c and AGE formation in the streptozotocin induced diabetic mice. As accumulation of AGEs is also implicated in development of insulin resistance.17 However, further studies are required to investigate the usefulness of pre-acetylation in prediabetic or insulin resistance condition to protect proteins from glycation.
Acknowledgements
This work financially supported by the grants from Council of Scientific and Industrial research (CSIR), New Delhi, India (CSC0111). MGJ thank Indian Council of Medical research (ICMR), India for research fellowship. KK thank CSIR, New Delhi for fellowship.
Notes and references
- T. J. Sims, L. M. Rasmussen, H. Oxlund and A. J. Bailey, Diabetologia, 1996, 39, 946–951 CrossRef CAS PubMed
 .
. - G. Basta, A. M. Schmidt and R. De Caterina, Cardiovasc. Res., 2004, 63, 582–592 CrossRef CAS PubMed .
- D. S. Raj, D. Choudhury, T. C. Welbourne and M. Levi, Am. J. Kidney Dis., 2000, 35, 365–380 CrossRef CAS PubMed .
- R. Castellani, M. A. Smith, G. L. Richey and G. Perry, Brain Res., 1996, 737, 195–200 CrossRef CAS PubMed .
- N. Sasaki, R. Fukatsu, K. Tsuzuki, Y. Hayashi, T. Yoshida, N. ujii, T. Koike, I. Wakayama, R. Yanagihara, R. Garruto, N. Amano and Z. Makita, Am. J. Pathol., 1998, 153, 1149–1155 CrossRef CAS PubMed .
- M. J. Kulkarni, A. M. Korwar, S. Mary, H. S. Bhonsle and A. P. Giri, Proteomics: Clin. Appl., 2013, 7, 155–170 CrossRef CAS PubMed .
- D. Hawkins, R. N. Pinckard and R. S. Farr, Science, 1968, 160, 780–781 CAS .
- R. Ajiboye and J. J. Harding, Exp. Eye Res., 1989, 49, 31–41 CrossRef CAS PubMed .
- M. S. Swamy and E. C. Abraham, Ophthalmol. Vis. Sci., 1989, 30, 1120–1126 CAS .
- R. B. Nahomi, T. Oya-Ito and R. H. Nagaraj, Biochim. Biophys. Acta, 2013, 1832, 195–203 CrossRef CAS PubMed .
- F. Caballero, E. Gerez, A. Batlle and E. Vazquez, Chem.-Biol. Interact., 2000, 126, 215–225 CrossRef CAS PubMed .
- F. Finamore, F. Priego-Capote, F. Gluck, A. Zufferey, P. Fontana and J. C. Sanchez, EuPa Open Proteomics, 2014, 3, 100–113 CrossRef CAS .
- J. Lei, Y. Zhou, D. Xie and Y. Zhang, J. Am. Chem. Soc., 2015, 137, 70–73 CrossRef CAS PubMed .
- Y. M. Kolekar, G. Vannuruswamy, S. B. Bansode, B. Santhakumari, H. V. Thulasiram and M. J. Kulkarni, RSC Adv., 2015, 5, 25051–25058 RSC .
- C. Patrono, N. Engl. J. Med., 1994, 330, 1287–1294 CrossRef CAS PubMed .
- D. Hawkins, R. N. Pinckard and R. S. Farr, Science, 1968, 160, 780–781 CAS .
- W. Cai, M. Ramdas, L. Zhu, X. Chen, G. E. Striker and H. Vlassara, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15888–15893 CrossRef CAS PubMed .
- B. Schmidt, F. Hölter, R. Berger and S. Jessel, Adv. Synth. Catal., 2010, 352, 2463–2473 CrossRef CAS .
- K. A. Mereish, H. Rosenberg and J. Cobby, J. Pharm. Sci., 1982, 71, 235–238 CrossRef CAS PubMed .
- H. S. Bhonsle, S. K. Singh, G. Srivastava, R. Boppana and M. J. Kulkarni, Protein Pept. Lett., 2008, 15, 663–667 CrossRef CAS PubMed .
- A. M. Korwar, G. Vannuruswamy, M. G. Jagadeeshaprasad, R. H. Jayaramaiah, S. Bhat, B. S. Regin, S. Ramaswamy, A. P. Giri, V. Mohan, M. Balasubramanyam and M. J. Kulkarni, Mol. Cell. Proteomics, 2015, 14, 2150–2159 CAS .
- H. S. Bhonsle, A. M. Korwar, S. K. Kesavan, S. D. Bhosale, S. B. Bansode and M. J. Kulkarni, Eur. J. Mass Spectrom., 2012, 18, 475–481 CrossRef CAS PubMed .
- S. K. Kesavan, S. Bhat, S. B. Golegaonkar, M. G. Jagadeeshaprasad, A. B. Deshmukh, H. S. Patil, S. D. Bhosale, M. L. Shaikh, H. V. Thulasiram, R. Boppana and M. J. Kulkarni, Sci. Rep., 2013, 15, 2941 Search PubMed .
- T.-L. Ho, Synth. Commun., 1977, 7, 393–395 CrossRef CAS .
- M. I. El Seoud, R. C. Vieira and O. A. El Seoud, J. Org. Chem., 1982, 47, 5137–5141 CrossRef CAS .
- M. Baarra and R. De Rossi', Can. J. Chem., 1991, 69, 1124–1130 CrossRef .
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11313c |
| ‡ Equally contributed. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 





