
Structural revision of aristol: a fresh look at the oxidative coupling of thymol under iodination conditions†
Niko S. Radulović*,
Miljana R. Đorđević and
Polina D. Blagojević
Department of Chemistry, Faculty of Science and Mathematics, University of Niš, Višegradska 33, 18000, Niš, Serbia. E-mail: nikoradulovic@yahoo.com; Fax: +381 18533014; Tel: +381 18533015
First published on 14th July 2016
Abstract
Aristol, an antiseptic drug prepared by iodination of thymol under alkaline conditions, has been on the market since the 1880s. Up until 1951, a myriad of (unlikely) structures were put forward, but none of them were in full agreement with the observed chemical and physical properties of aristol. Today, according to most pharmacopoeias and commercial sources, aristol represents an iodooxybiphenyl derivative; the origin of this structure is unknown. Motivated by these facts, we decided to elucidate the structure of aristol using modern chromatographic and extensive spectrometric techniques (NMR/FTIR/MS/UV), combined with chemical transformations and quantum mechanical calculations. Based on our findings, it was revealed that aristol does not represent a single chemical entity but a complex mixture of a large number of structurally closely related iodinated dehydrodithymol molecules together with (di)iodothymols and unreacted thymol. Five products of oxidative coupling (CAr–CAr, CAr–O–CAr and CAr–CH2–CAr) were successfully isolated in a pure state and fully spectrally characterized: 3,3′-diiodo-5,5′-diisopropyl-2,2′-dimethyl-[1,1′-biphenyl]-4,4′-diol, 5,5′-diiodo-3,3′-diisopropyl-6,6′-dimethyl-[1,1′-biphenyl]-2,2′-diol, 2-iodo-4-(4-iodo-2-isopropyl-5-methylphenoxy)-6-isopropyl-3-methylphenol, 4-(5-hydroxy-2-iodo-4-isopropylbenzyl)-2-isopropyl-5-methylphenol, and 3-(4-hydroxy-5-isopropyl-2-methylbenzyl)-2,4-diiodo-6-isopropylphenol. A lateral meta-coupling (with respect to the phenoxy-radical), CAr–CH2–CAr, previously never reported for oxidative coupling of phenols, was firmly established in two aristol constituents. This provides evidence for a mechanism that involves benzyl radicals unknown to form under these conditions. An additional 16 aristol constituents were identified by the use of a QSPR model (readily available structure-derived descriptors used to predict gas chromatography retention data), in combination with mass spectrometry, directly from the aristol matrix without preparative chromatography. The herein presented results urge a revision of the currently accepted formula (composition) of aristol in both primary and secondary literature.
Introduction
Aristol, an iodo derivative of thymol, is considered to be an excellent and prompt antiseptic.1,2 It has been used for the treatment of various skin conditions (varicose ulcers, psoriasis, skin abscess), gonorrhoea, lymphadenitis, periostitis, etc.1–3 Aristol has also been used, in dental medicine (gangrenous pulps, antisepticising of root-canals, etc.), in all cases when ordinary antiseptics are indicated.1,2,4 When compared to iodoform, it is free from irritant action upon unbroken skin, produces no toxic known effects and has no unpleasant odor.1Aristol has been on the market for about 130 years, sold under a number of trade names: diiododithymol, annidalin, thymotol, iodothymol, thymodin, thymol iodide and others;5 according to recent recommendations, aristol should be placed on the list of bulk substances allowed for compounding under section 503A of the US Federal Food, Drug, and Cosmetic Act.2 There is a common misbelief that aristol represents a chemically well-characterized substance.2,3 Its chemistry and identity (structure) had been investigated intensely in the period of 1889 to 1950.6–13 Despite having about 100 years of literature history, the structure of the substance(s) named aristol is elusive still today, and the mechanism of its formation is not satisfactorily explained.6–13 Although the molecular formula for dithymol diiodide is usually given as (C10H12IO)2, with a molecular weight of 550, there is still uncertainty as to how the iodine and thymol moieties are mutually bound.13
Several classical (degradative and synthetic) reports on the structure of aristol appeared in the mentioned period, but no studies that draw their conclusions from modern spectroscopic techniques exist. In 1889, Messinger and Vortmann proposed structure 1 for the alleged “red aristol” and 2 for “yellow aristol”.6 They perceived aristol as hypoiodous esters of iodized phenols (an iodoxyl derivative), but failed to give proof of their view. Seven years later, after repeating the synthesis of aristol, Urban suggested structure 3, but the necessary information supporting the structure was not given in the paper itself (Fig. 1).7
 | ||
| Fig. 1 Previously proposed structures of aristol.2,6–12 | ||
Dannenberg (1903) also proposed structures for “red and yellow aristols” (4 and 5, respectively).8 Bougault (1918) put forward formula 6 in which the iodine was connected directly to the aromatic core of thymol and the two thymol structural fragments were additionally linked through a peroxide bridge.9 Moles and Marquina (1919) gave a symmetrical structure 7 with free phenolic groups, for the first time.10 Sanna and Zucca (1951) believed that aristol is an anthracene derivative 8.11 Today, most pharmacopoeias and commercial sources (for example, confer the site of Sigma-Aldrich) list it as 4,4′-bis(iodooxy)-2,2′-dimethyl-5,5′-bis(1-methylethyl)-1,1′-biphenyl, although there is no conclusive proof that supports structure 9.2,12
The challenges in the structural elucidation of aristol might have their origin in the fact that it most probably does not represent a single compound, but a mixture of related ones. Much of the circumstantial evidence, accumulated by the classical chemical analyses (solubility, reactivity and certain physical constants), based on which structural assignments were made, might have varied from one batch of aristol to another due to varying relative amounts of the constituting compounds.
Thus, there is a general absence of evidence for any of the previously proposed structures of aristol. Also, the existence of molecules with many of the proposed structures (1–6, 9), including the currently accepted one (9) by a number of pharmacopoeias, is highly improbable (unstable hypoiodite compounds, unlikely tautomeric forms etc.). Motivated by this, we decided to determine the structure of aristol by employing the spectroscopic tools of a modern chemist and to verify whether it represents a single compound or a mixture. By doing so, we could possibly enable future pharmacological studies, i.e. the location of active principles of aristol. We envisaged this study to start by a chromatographic separation of the potential constituents of the multicompound drug called collectively “aristol”, prepared by iodination of thymol under alkaline conditions.13 Even a simple chromatography on silica gel (dry flash) of the reaction mixture, obtained in a straightforward iodination of thymol under alkaline conditions, yielded several pure products of oxidative coupling. The structures of the isolated compounds were deduced from detailed analyses of their 1D- and 2D-NMR spectra, along with other spectral data (MS, IR and UV). Thus, in this work, for the first time, based on extensive spectral data, we report the structures of five novel compounds that constitute the commercial drug “aristol”. Among the identified compounds, two compounds represent products of an unreported oxidative lateral meta-coupling (with respect to the phenoxy-radical) of any phenol. The mechanism of their formation was discussed in the light of the existence of benzyl radicals that were unknown to form under these conditions.
Results and discussion
Structural elucidation
Iodination of thymol, under alkaline conditions, was performed as described by Woollett,13 in an attempt to obtain a substance referred to as “aristol”. The resulting crude reaction product was then subjected to chromatographic separation. A gradient “dry flash” chromatography on a SiO2 column, using mixtures of hexane and diethyl ether of increasing polarity as the eluents, yielded two pure compounds (16 and 17, Fig. 2). | ||
| Fig. 2 Fully spectrally characterized constituents of aristol; the numbering scheme, used in the NMR discussion is based on the p-menthane core. | ||
Re-chromatography of one of the fractions (fraction I, see Experimental section) gave 6 additional pure compounds. The structural elucidation of the isolated purified aristol constituents – 5 oxidative coupling products (7, 14–17; Fig. 2), diiodothymol (13), two monoiodothymol regioisomers (11, 12) and unreacted thymol (10) – was achieved by a combination of NMR (1D: 1H and 13C NMR, and selective 1H homodecoupling experiments; 2D: HSQC, HMBC, 1H–1H-COSY and NOESY experiments), HRMS, FTIR and UV spectral techniques, as well as chemical transformations (methylation). The NMR data are listed in Table 1 (for 10–13) and 2 (7, 14–17) (carbon and proton atom numbering schemes, shown in Fig. 2, were based on the p-menthane parent structure to allow an easier comparison among the different coupling products). Herein, for the first time, we provide unambiguous proof on the structure of five coupling products (7, 14–17) identified as aristol constituents which, according to a search of the CAS database (SciFinder, accessed on May 2016), represent novel compounds. Interestingly, the ortho-regioisomer of monoiodothymol (11) was not registered in the CAS database. Similarly, compound 7, although its structure was reported in Moles and Marquina,10 was also not included in this database. In the paragraphs that follow, we first give the details regarding the structural elucidation of the mentioned aristol constituents.
| Position | 10 | 11 | 12 | 13 | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, mult. | δH (J in Hz) | δC, mult. | δH (J in Hz) | δC, mult. | δH (J in Hz) | δC, mult. | |
| 1 | 136.6 C | 139.2 C | 139.5 C | 140.2 C | ||||
| 2 | 6.57, dqd (1.7, 0.6, 0.3) | 116.0 CH | 94.5 C | 6.66 (pseudo q) | 116.9 CH | 93.4 C | ||
| 3 | 152.4 C | 151.6 C | 152.7 C | 152.3 C | ||||
| 4 | 131.3 C | 132.2 C | 134.4 C | 134.5 C | ||||
| 5 | 7.08, dd (7.8, 0.3) | 126.2 CH | 7.05, d (7.7) | 126.1 CH | 7.54, s | 136.5 CH | 7.57, s | 136.8 CH |
| 6 | 6.73, dqd (7.8, 1.7, 0.5) | 121.7 CH | 6.81, d (7.7) | 121.8 CH | 90.2 C | 88.1 C | ||
| 7 | 2.27, br s | 20.9 CH3 | 2.41, s | 28.4 CH3 | 2.32, br s | 27.5 CH3 | 2.72, s | 35.2 CH3 |
| 8 | 3.16, septuplet (6.9) | 27.5 CH | 3.28, septuplet (6.9) | 26.7 CH | 3.10, septuplet (6.9) | 26.6 CH | 3.23, septuplet (6.9) | 28.3 CH |
| 9 | 1.24, d (6.9) | 22.7 CH3 | 1.20, d (6.9) | 22.5 CH3 | 1.22, d (6.9) | 22.5 CH3 | 1.21, d (6.9) | 22.3 CH3 |
| 10 | 1.24, d (6.9) | 22.7 CH3 | 1.20, d (6.9) | 22.5 CH3 | 1.22, d (6.9) | 22.5 CH3 | 1.21, d (6.9) | 22.3 CH3 |
C3–O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) |
4.70, br s | 5.48, br s | 4.77, br s | 5.51, br s | ||||
In order to facilitate the interpretation of NMR spectral data of the coupling products 7 and 14–17, the NMR data of thymol and its mono- and diiodo derivatives were fully assigned. Strangely, to the best of our knowledge, a complete spin analysis of 1H multiplets of thymol (10) does not exist in the literature. A number of long-range couplings of protons (for example, H2 coupled with H6 (4J = 1.7 Hz), H5 (5J = 0.3 Hz) and H7 (4J = 0.6 Hz)) were disclosed by a series of selective 1H homonuclear decoupling experiments. The total assignation of the resonances from the 1H and 13C NMR spectra (Table 1) was aided by 2D NMR (1H–1H COSY, HMBC, HSQC and NOESY).
High resolution mass spectrometry (HRMS) and elemental analysis of compounds 11 and 12 suggested these were two (different) monoiodo derivatives of thymol: molecular ions for these compounds appeared at m/z 276.0001 (Δ = −0.0010 amu) and m/z 276.0000 (Δ = −0.0011 amu), respectively (this value corresponded to the molecular formula C10H13IO). The position of the iodine atom in each of them was straightforwardly inferred from their NMR data. In the case of o-iodothymol (11), 6 resonances were observed in the corresponding proton NMR spectra. The values of H8–H10 chemical shifts were slightly downfielded compared to those of thymol (Table 1), while their general appearance (multiplicity) remained the same. In addition to the lack of the signal corresponding to H2, the most pronounced noted difference was in the value of the chemical shift of the phenolic hydrogen atom (δ 5.48 and 4.70 ppm, for 11 and 10, respectively) suggesting a close proximity of this OH to an electronegative atom (I). The position of the iodine atom was also evident from the significant upfield shift of the C2 carbon atom resonance when compared to that of thymol (Δδ = −21.5 ppm). The general C–H connectivity and the assignation of all 1H- and 13C-NMR signals was based on the data from HMBC, HSQC and NOESY spectra (Fig. S1, ESI†). It is worth mentioning that the HMBC spectrum of this compound displayed an interaction between C2(–I) and H5 over 4 bonds suggesting a relatively large value of the long-range C–H coupling (two coupling pathways available) between these nuclei, being in the range of values of 2- or 3-bond C–H couplings.
The spectral data of 12 and 13 were also missing in the literature. In an analogous way as for 11, the herein recorded spectral data of compounds 12 and 13 allowed their structures to be established as p-iodothymol (12) and o,p-diiodothymol (13), Fig. 2, and their assigned NMR data are presented in Table 1. The position of the iodine atom(s) was apparent from the values of the chemical shifts of C6 in 12 (Δδ = −31.5 ppm), and C2 and C6 in 13 (Δδ = −22.6 and −33.6 ppm, respectively) when compared to those of thymol. As expected, the chemical shifts of their phenolic protons were shifted to lower field. The deshielding (around 0.8 ppm; Fig. S1†) of the OH in diiodothymol was much more pronounced and comparable in value to that noted in o-iodothymol, a piece of information that turned out to be useful in the structural elucidation of the coupling products (7, 14–17).
First clues on the identity of compound 14 came from HRMS. The molecular ion ([M]+; this was also the base peak in the MS of 14) was observed at m/z 549.9880 (Δ = +0.0014 amu), suggesting the molecular formula C20H24I2O2; the ratio between the intensities (Int) of [M]+ and [M + 1]+ ions, Int(M+)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Int(M + 1) = 100:22, also indicated that 14 was comprised of 20 carbon atoms. Thus, 14 was assumed to be a diiodo derivative of an oxidative coupling product of two molecules of thymol. The second and the third most intense peaks in the MS of 14 were those with m/z 535 and 260. These corresponded to [M − CH3]+ and [M − CH3 − C10H12IO]+ ions, respectively (Fig. S2†). Hence, 14 does not seem to bear two iodine atoms on the same thymol core (the loss of the fragment C10H12IO in a single step would not be possible in that case). The presence of a phenolic group and a substituted benzene ring in 14 was concluded from the observed IR bands at 3473 (phenol O–H stretch) and 1596 cm−1 (aromatic C
:Int(M + 1) = 100:22, also indicated that 14 was comprised of 20 carbon atoms. Thus, 14 was assumed to be a diiodo derivative of an oxidative coupling product of two molecules of thymol. The second and the third most intense peaks in the MS of 14 were those with m/z 535 and 260. These corresponded to [M − CH3]+ and [M − CH3 − C10H12IO]+ ions, respectively (Fig. S2†). Hence, 14 does not seem to bear two iodine atoms on the same thymol core (the loss of the fragment C10H12IO in a single step would not be possible in that case). The presence of a phenolic group and a substituted benzene ring in 14 was concluded from the observed IR bands at 3473 (phenol O–H stretch) and 1596 cm−1 (aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bending), and UV adsorptions at λmax 208.0 and 282.5 nm (characteristic for a substituted electron-rich aromatic ring). After a methylation of a sample of 14, a product with 28 amu higher molecular weight was obtained indicating that there were two free phenolic groups present in the molecule.
C bending), and UV adsorptions at λmax 208.0 and 282.5 nm (characteristic for a substituted electron-rich aromatic ring). After a methylation of a sample of 14, a product with 28 amu higher molecular weight was obtained indicating that there were two free phenolic groups present in the molecule.
There were only 6 different resonances observable in the 1H NMR spectra of 14 (Table 2), including only one (δ 6.89, singlet) corresponding to a proton directly attached to an aromatic ring (Ar–H). Thus, we hypothesized 14 to be a symmetrical compound, comprised of two identical iodothymol fragments with directly (CAr–CAr) coupled benzene rings. This was further confirmed by the number (10) of observed 13C NMR signals, 5 of which were not attached to a proton. The observed 1H and 13C chemical shifts resembled those of o-iodothymol (11), with H6 missing and H5 reduced to a singlet, as mentioned above, suggesting para C–C coupling (with respect to the OH) of two o-iodothymol units. The position of the iodine atom within this unit was confirmed by the existence of a three-bond HMBC correlation observed between the broad singlet at δ 5.53, assigned to the phenolic proton, and the resonance of the carbon (C2) attached to I (Fig. 3); this carbon also showed a cross-peak with the protons at 2.15 ppm (H7, methyl group).
| Position | 14 | 7 | 15 | 16 | 17 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC, mult. | δH (J in Hz) | δC, mult. | δH (J in Hz) | δC, mult. | δH (J in Hz) | δC, mult. | δH (J in Hz) | δC, mult. | |
| 1 | 136.8 C | 138.1 C | 130.9 C | 141.6 C | 142.3 C | |||||
| 2 | 95.7 C | 120.7 C | 95.2 C | 6.09, s | 116.0 CH | 94.4 C | ||||
| 3 | 150.9 C | 151.2 C | 148.8 C | 153.2 C | 152.5 C | |||||
| 4 | 132.0 C | 135.8 C | 133.5 C | 134.8 C | 135.2 C | |||||
| 5 | 6.89, s | 128.1 CH | 7.71, s | 137.7 CH | 6.75, s | 118.9 CH | 7.59, s | 136.7 CH | 7.66, s | 137.2 CH |
| 6 | 134.8 C | 91.3 C | 146.4 C | 90.1 C | 89.7 C | |||||
| 7 | 2.15 s | 26.4 CH3 | 2.06, s | 25.2 CH3 | 2.28, s | 22.0 CH3 | 3.80, s | 44.0 CH2 | 4.26, s | 49.3 CH2 |
| 8 | 3.33, septuplet (7.0) | 28.4 CH | 3.22, septuplet (6.9) | 27.2 CH | 3.29, septuplet (6.9) | 28.6 CH | 3.10, septuplet (7.0) | 26.7 CH | 3.30, septuplet (6.9) | 28.4 CH |
| 9 | 1.22, d (7.0) | 22.5 CH3 | 1.29, d (6.9) | 22.3 CH3 | 1.18, d (6.9) | 22.6 CH3 | 1.22, d (7.0) | 22.4 CH3 | 1.26, d (6.9) | 22.3 CH3 |
| 10 | 1.24, d (7.0) | 22.6 CH3 | 1.29, d (6.9) | 22.3 CH3 | 1.18, d (6.9) | 22.6 CH3 | 1.22, d (7.0) | 22.4 CH3 | 1.26, d (6.9) | 22.3 CH3 |
| C3–O |
5.53, br s | 4.68, s | 5.38, br s | 4.73, br s | 5.61, br s | |||||
| 1′ | 136.8 C | 138.1 C | 139.7 C | 135.4 C | 127.3 C | |||||
| 2′ | 95.7 C | 120.7 C | 6.31, s | 116.0 CH | 6.61, s | 117.2 CH | 6.63, s | 117.1 CH | ||
| 3′ | 150.9 C | 151.2 C | 155.9 C | 151.4 C | 150.8 C | |||||
| 4′ | 132.0 C | 135.8 C | 137.2 C | 131.8 C | 131.3 C | |||||
| 5′ | 6.89, s | 128.1 CH | 7.71, s | 137.7 CH | 7.64, s | 136.8 CH | 6.89, s | 128.5 CH | 6.29, s | 124.8 CH |
| 6′ | 134.8 C | 91.3 C | 92.0 C | 129.9 C | 134.5 C | |||||
| 7′ | 2.15, s | 26.4 CH3 | 2.06, s | 25.2 CH3 | 2.25, s | 27.8 CH3 | 2.07, s | 19.1 CH3 | 2.33, s | 19.5 CH3 |
| 8′ | 3.33, septuplet (7.0) | 28.4 CH | 3.22, septuplet (6.9) | 27.2 CH | 3.34, septuplet (6.9) | 26.8 CH | 3.15, septuplet (7.0) | 26.8 CH | 3.02, septuplet (6.9) | 26.8 CH |
| 9′ | 1.22, d (6.9) | 22.5 CH3 | 1.29, d (6.9) | 22.3 CH3 | 1.27, d (6.9) | 22.7 CH3 | 1.22, d (7.0) | 22.7 CH3 | 1.14, d (6.9) | 22.5 CH3 |
| 10′ | 1.24, d (6.9) | 22.6 CH3 | 1.29, d (6.9) | 22.3 CH3 | 1.27, d (6.9) | 22.7 CH3 | 1.22, d (7.0) | 22.7 CH3 | 1.14, d (6.9) | 22.5 CH3 |
| C3′-O |
5.53, s | 4.68, s | 4.87, br s | 4.82, br s | ||||||
 | ||
| Fig. 3 HMBC and NOESY interactions crucial for the structural elucidation of compounds 7, 14–17. | ||
A NOESY cross-peak between H7 methyl protons from one of the aromatic cores and H5′ from the other core (i.e. H5 and H7′) corroborated the coupling site (C6–C6′). An additional piece of information that substantiated this mode of coupling was the upfield shift (when compared to the corresponding signal in 11) of the methyl protons (H7) due to the anisotropic effect of a benzene ring on a nucleus (protons) placed above the plane of the ring (2.15, 2.41 ppm, for H7 in 14 and 11, respectively; Fig. S3†). Based on all of the data above, the structure of compound 14, a symmetrical didehydrodithymol derivative, was deduced to be that of 3,3′-diiodo-5,5′-diisopropyl-2,2′-dimethyl-[1,1′-biphenyl]-4,4′-diol.
The isopropyl CH3 groups in thymol, monoiodothymols and diiodothymol are enantiotopic and accordingly only one signal was observed for these CH3 groups in their 1H and 13C NMR spectra. The existence of a chiral axis in compound 14, due to hindered rotation abound the Ar–Ar bond, makes these methyl groups become mutually diastereotopic (within the isopropyl group), i.e. there are 4 different stereoisomers (two pairs of enantiomers) that could be derived from the substitution of one of the CH3 groups with, for example, a CD3 group. And indeed, in 14, there were two doublets in the 1H NMR and two 13C signals observed corresponding to these CH3 groups. This obvious consequence of atropisomerism corroborates the proposed structure of 14.
The molecular ion [M]+ (m/z 549.9847, Δ = −0.0019 amu) of compound 7 corresponded to the molecular formula C20H24I2O2. The observed MS fragmentation pattern, IR and UV spectra were almost identical to that of 14. Thus, we were probably dealing with an isomer of 14. Just as in the case of compound 14, the number of resonances of 1H (a proton directly attached to an aromatic core, Ar–H, and the signals of the following groups: CH3Ar, CH(CH3)2 and a phenolic OH) and 13C (10 signals) nuclei noted in the respective NMR spectra, suggested compound 7 to be a symmetrical dehydrodimer, as well, comprised of two pentasubstituted benzene rings. Methylation of 7 led to an increase in molecular mass of 28 amu, implying the presence of two free phenolic OH groups. The values of the chemical shift of the Ar–H (δ 7.71) and OH (δ 4.68) protons were closest in value to that of the corresponding p-iodothymol protons (12; δ 7.54; δ 4.77, respectively, Tables 1 and 2, Fig. S1†), indicating that identical p-iodothymol moieties were directly (CAr–CAr) coupled via C2/C2′ atoms in compound 7. The existence of a NOE correlation between the phenolic proton (OH) from one and the H7′ methyl protons from the other benzene core (Fig. 3) provided the final evidence on the coupling mode of the benzene rings in 7; no such interaction was possible within a single aromatic unit. This together with other important NOESY and HMBC interactions (e.g. H5–C6, C2–H7 or C2–OH) observed for compound 7, shown in Fig. 3, allowed us to assign compound 7 with the structure of 5,5′-diiodo-3,3′-diisopropyl-6,6′-dimethyl-[1,1′-biphenyl]-2,2′-diol (7). Again, due to the anisotropy of the benzene ring, the signal of the CH3Ar group (δ 2.06) was shifted upfield by about 0.26 ppm with respect to that of the “monomeric” counterpart, p-iodothymol (12). Similarly, as for 14, the diastereotopic isopropyl CH3 groups were found to display different chemical shifts.
We isolated yet another isomer of compounds 7 and 14, compound 15 (C20H24I2O2; molecular ion at m/z 549.9901, Δ = +0.0035). Although its IR and UV spectra (these revealed the presence of an aromatic (benzene) core and/or OH group), were comparable to the spectra of the two mentioned compounds, its MS fragmentation pattern differed from that of 7 and 14. The presence of only one free phenolic group could be deduced from a methylation experiment (a single CH3 was introduced into 15). A much greater number of resonances observable in the 1H NMR (10 in total; these corresponded to 2 different Me and iPr-groups, 3 different Ar–H protons and one exchangeable OH proton) and 13C NMR spectra (18 in total; the signals of iPr carbons were isochronous) implied that 15 was a coupling product of either two non-identical iodothymol moieties or two identical ones but non-symmetrically coupled. The values of the chemical shifts and the multiplicities of the Ar–H resonances (singlets at δ 6.31, 6.75 and 7.64) suggested that there were no Ar–H protons mutually ortho and that there is only a single proton that was ortho to an iodine atom (δ 7.64, H5′; Fig. S1;† p-iodothymol fragment). An HMBC correlation between C2, δ 95.2 (this resonance was upfielded in respect to the corresponding one in thymol due to the fact that this C was directly bonded to iodine), and the H-atom from C3–O, δ 5.38, revealed that an o-iodothymol unit carried the free OH group (the high value of C3–O chemical shift was in agreement with this assumption, Fig. S1†). Three sp2 carbon atoms (δ 146.4, 148.8 and 155.9, for C6, C3 and C3′, respectively) were directly linked to a strongly deshielding oxygen atom. As this is only possible if the two aromatic cores were coupled via an oxygen atom (CAr–O–CAr), we concluded that 15 is a coupling product of o- and p-iodothymols via the O atom from p-iodothymol. This assumption was further corroborated by the presence of HMBC correlations between H2′–C6′ (δ 6.31 and 92.0 ppm, respectively), H5–C6 and H5–C3 (δ 6.75, 146.4 and 148.8 ppm, respectively). In addition, there was a NOESY interaction between the signals from the two different aryl moieties, at δ 6.31 (H2′) and δ 2.28 (H7), demonstrating that these protons were spatially close, i.e. the proposed mode of coupling (Fig. 3). Thus, compound 15 was deduced to be 2-iodo-4-(4-iodo-2-isopropyl-5-methylphenoxy)-6-isopropyl-3-methylphenol.
Based on the molecular formula of compound 16, it appeared to be a monoiodo derivative of an isomer of dehydrodithymol (C20H25IO2; according to HRMS, [M]+ at m/z 424.0871, Δ = −0.0028 amu), with two free OH-groups (a broad band at 3365 cm−1 was observable in its IR, and two exchangeable protons at 4.73 and 4.87 ppm that were not in the close neighbourhood of an I-atom, Fig. S1†). The coupling mode of aromatic cores in 16 was inferred from 1D and 2D NMR. Unexpectedly, 1H NMR spectrum showed that in addition to a single CH3Ar (a singlet at δ 2.07, with an integral 3), the molecule of 16 had one CH2-group (a singlet at δ 3.80, with an integral 2). DEPT-135 experiment confirmed the existence of a methylene group with a rather high value of its chemical shift (δ 44.0), that implied that the CH2 was bridging the two aromatic rings (the shifts of the corresponding methyl protons from 7, 10–16 were in the range 19.1–35.2 ppm). Based on this, compound 16 was a product of a rather unusual benzyl (CAr–CH2–CAr) coupling of thymol and an iodothymol isomer. As all of the Ar–H protons (Table 2) appeared as singlets, it seems that C7 from a p-iodothymol fragment was connected to C6′ (para position in respect to OH group) of the otherwise unsubstituted thymol moiety. Any other CAr–CH2–CAr coupling mode would yield a product with a pair of mutually ortho Ar–H protons. The observed HMBC and NOESY experiments were in agreement with this hypothesis (Fig. 3). In the HMBC spectrum, methylene H7 correlated with C1 (141.6 ppm), C2 (116.0 ppm), C6 (90.1 ppm; directly attached to the I-atom), C1′ (135.4 ppm), C5′ (128.5 ppm) and C6′ (129.9 ppm) (Fig. 3). NOESY cross-peaks between H7–H5′, H7–H7′ and H7–H2 further confirmed that the structure of 16 corresponds to 4-(5-hydroxy-2-iodo-4-isopropylbenzyl)-2-isopropyl-5-methylphenol (Fig. 2). The values of C3–O and C3′–O chemical shifts (δ 4.73 and 4.87 ppm) followed the same trend as the one established for 7, 10–15 (Fig. S1†).
The final coupling product isolated in pure state was, again, an isomer of 7, 14 and 15. According to 1H NMR and DEPT-135 spectra, compound 17 was also recognized as a CAr–CH2–CAr coupling product of thymol (methylene 1H and 13C resonances appeared at δ 49.3 (C7), i.e. δ 4.26 ppm (H7); the signals of all Ar–H were singlets), but with two iodine atoms in its structure (C20H24I2O2; according to HRMS, [M]+ at m/z 550.9845, Δ = −0.0021). There was a long-range C–H correlation between the methylene protons and carbon atoms at δ 94.4 and 89.7 (low value of their chemical shifts indicated that these were directly bonded to I-atoms). At the same time, no HMBC correlations were observable between the Ar–CH3 and the mentioned iodinated C-atoms. Thus, it appeared that both iodine atoms were attached to the same aromatic core. The occurrence of HMBC cross-peaks between the signals at δ 7.66–89.7 (H5–C6), 7.66–94.4 (H5–C2), 7.66–152.5 (H5–C3), 5.61–152.5 (C3–O–C3) or 5.61–94.4 ppm (C3–O–C2) was in line with the mentioned assumption. The NOESY correlation between the H-atoms at δ 4.26 (H7) and 6.29 (H5′) and between H2′ and C3′–O (δ 4.82) confirmed the coupling mode present in compound 17. All of these data led to the assignment of the structure of 17 as that of 3-(4-hydroxy-5-isopropyl-2-methylbenzyl)-2,4-diiodo-6-isopropylphenol.
To further corroborate the structural assignment of aristol constituents, we have optimized geometries of purified iodinated products 7, 11–17 and theoretically calculated chemical shifts for both 1H and 13C nuclei (GIAO method; B3LYP/6-31G++(p,d) level of theory).14 In order to be able to evaluate the accuracy of the applied computational method, the same was done for thymol (10). The calculated values of 1H/13C NMR shifts of thymol were in good agreement with those experimentally observed (correlation coefficients, R2, between these values were 0.99 and 0.97 for 13C and 1H respectively), although slightly overestimated (for ca. 10 ppm in the case of carbon, i.e. 0.2 ppm for proton shifts). This was true for the rest of the compounds (correlation coefficients, R2, were higher than 0.93; the highest differences between the calculated and the experimental chemical shifts were observed for polar (OH) protons and C-atoms directly attached to an iodine atom). These values could be used as yet another confirmation of the substitution pattern and/or coupling mode of compounds 11–17. For example, both the calculated (without scaling) and experimental values of 1H NMR chemical shifts (of “aromatic” protons) for compounds 10–13 and 16 are given in parallel in Table S1.† In the optimized geometry of 16 (energetic minimum), H2 was placed right above the ring of the other aromatic core, i.e. within its shielding cone, Fig. S3.† However, due to the (at least partial) conformational freedom along C1–C7–C6′ backbone, the geometries in which the H2 nucleus is not under the (anisotropic) shielding influence of the other Ar ring are also possible. Thus, it is not surprising that the experimentally observed shielding of H2 in 16 (when δ-H2 in 16 and 10 or 12 are compared) was lower than the calculated one, Table S1.† For all other aromatic core protons (including the H5′ proton that also might fall within the Ar deshielding cone, Fig. S3†), the difference between the calculated and the experimental values was less than 0.3 ppm (without scaling). Similarities/dissimilarities in the calculated values for the corresponding nuclei from 10–13 and 16 (these are in line with the experimentally observed ones) confirm that compound 16 is comprised of the monomeric units of thymol, 10, and p-iodothymol, 12, bridged by a methylene group.
Compounds 7, 11–17 differed (mutually and from other compounds present in the remainder of the mixture, i.e. “aristol”) both in their polarity and accessibility of their phenolic groups (steric hindrance imparted by the proximity of an iodine atom and/or alkyl group) to intermolecular hydrogen bonding with the SiO2-stationary phase. The order of elution (with mixtures of hexane and diethyl ether; cf. Fig. S4†) from the SiO2 column provided yet another proof of the above-established structures of these compounds. An ortho iodine atom with respect to the phenolic group of thymol imparted a steric impediment to hydrogen bonding with the silanol (SiOH) groups from the stationary phase. As a result of this, compounds 11, 13–15 were eluted first from the SiO2 column. The phenolic OH became more acidic with the introduction of iodine para to this functionality, consequently making the hydrogen bond with the stationary phase stronger; i.e. longer chromatographic retention of compounds with this regiochemistry, in comparison to the non-iodinated cores and ortho-iodinated ones, was observed. Compound 7, although having two phenolic groups with para iodine atoms to them was eluted before thymol itself due to another kind of steric hindrance of hydrogen bonding caused by the two aromatic cores coupled in the vicinity of the two OH groups. Naturally, two accessible OH groups (as in 16) were found to make the molecule more prone to hydrogen bonding than those with only one such group (10, 12 and 17).
To summarize, the chromatographic separation of aristol, i.e. a mixture of compounds obtained by an alkaline iodination of thymol, afforded eight pure iodinated compounds (7, 11–17); five of these (one mono- and four diiododidehydrodithymols) represented new compounds formed by an apparent oxidative coupling of iodothymols (three different coupling modes: CAr–O–CAr, CAr–CH2–CAr and direct CAr–CAr). However, as evidenced by TLC, some other aristol constituents remained unidentified in a number of inseparable chromatographic fractions (see Experimental part). In order to try to possibly reveal the identity of some additional constituents of aristol, the original aristol mixture and the inseparable fractions were subjected to GC-MS analyses. The total ion current (TIC) chromatogram (Fig. 4) of the primary mixture revealed that alongside compounds 7, 10–17, there was a large number of additional compounds present in aristol, with relative amounts up to several percent of the total peak-areas of the detected compounds; relative abundances of some of these were comparable or even higher than that of 7, 10–17. The similarities of MS fragmentation patterns, molecular masses and retention indices of these unidentified aristol constituents (the search of the available MS databases gave no positive matches) with those of 7, 10–17 suggested that these unidentified compounds are related to them, i.e. that they might also be products of oxidative coupling of thymol and/or iodinated thymols. In order to possibly deduce the identity of the mentioned additional aristol constituents, we first turned to a consideration of the likely reaction mechanism of this iodination–oxidative coupling of thymol (we anticipated this to narrow down the number of expected products).
 | ||
| Fig. 4 Total ion current chromatogram of a sample of aristol prepared by an alkaline iodination of thymol. | ||
Possible mechanism of iodination–oxidative coupling of thymol
Generally speaking, on oxidation, electron-rich aryls (e.g. phenols, alkyl aryl ethers and the like) can undergo oxidative coupling reactions forming aryl–aryl bonds. The dehydrodimerization is a twofold C–H transformation which can proceed by several reaction pathways depending on the nature of the substrate and reagent mixture.15,16 For example, the loss of one proton and one electron from a molecule of thymol (possibly via intermediate b, Fig. 5 that can form upon treatment of 10 with iodine under alkaline conditions) would initially give an aryloxy radical (b1); the same should be true for o- or p-iodothymol derivatives 11 and 12. The unpaired electron is delocalized over the aromatic core (resonance structures b2–b4), rendering C2 and C6 (or even C4) as additional positions available for coupling. A recombination of these C-centred radicals would result in the formation of dimers that, in the case of b2 and b3, should tautomerize rapidly in a protic solvent (water) to the stable aromatic Ar–Ar coupling products e–g (or their iodinated analogues). The formation of compounds 7 and 14 could be explained in this way. Similarly, a recombination of O- (b1) with C-centred radicals (b2, b3) may yield Ar–O–Ar products c and d (or their iodinated analogues; Fig. 5). Compound 15 most likely formed via this coupling route. | ||
| Fig. 5 Possible mechanism of formation of 7 (mode g), 14 (mode e), 15 (mode c). | ||
Whether a coupling of iodinated radicals took place or that the iodine atom(s) was(were) subsequently introduced into the already coupled thymol moieties cannot be concluded straightforwardly. However, based on the ratio of ortho- and para-monoiodothymol observable in the reaction mixture, where ortho-iodination was hampered by sterics, all ortho-substituted (with respect to the OH group) dithymol derivatives appear to have their ortho-iodine introduced later on during the reaction course, i.e. when no other position for iodination was available. Once one iodine atom was already in, the introduction of the second one appeared to be much less problematic (based on the amount of diiodothymol present in aristol). In the case of compound 15, the iodine para to the ether bridge most probably originated from the monomeric p-iodothymol precursor due to the lesser nucleophilicity of the aryloxy substituted core when compared to the phenolate one. Also, since 15 was the most abundant constituent of aristol it is likely that it formed from the most abundant monoiodo derivative of thymol. However, a mixed (iodination prior and after the coupling step) origin of these iodinated coupling products cannot be ruled out.
Unlike 7, 14 and 15, compounds 16 and 17 seem to be products of an oxidative lateral meta-coupling (with respect to the phenoxy-radical).
The mechanism of formation of 16 and 17 (see Fig. 6) probably includes an initial formation of benzylic radicals that recombine with b2-type radicals to give CAr–CH2–CAr products (Fig. 6). The mechanistic course remains obscure for these particular meta-coupling products, while, according to some authors, primary oxidation at the phenolic oxygen followed by hydrogen migration from a benzylic site seems plausible for the formation of an ortho-substituted benzylic radical.15 The calculated values (see Experimental section) of bond dissociation energies (D0) for thymol and mono/diiodothymols benzyl C–H bonds – these can be used as a measure of the stability of the corresponding benzyl radicals – were relatively low, mutually comparable (366–368 kJ mol−1) and in agreement with both the previously calculated or experimentally determined for similar C-centred radical species.17,18 These values permit one to speculate on the existence of such meta-oriented benzylic radicals, previously unknown to form under oxidative coupling conditions, as intermediates in the mechanism of formation of 16 and 17.
 | ||
| Fig. 6 Possible mechanism of formation of 16 and 17. | ||
To the best of our knowledge, previous reports on lateral benzylic oxidative coupling of alkyl phenols (including iodinated ones) seem to be limited to the cases when the benzylic radical was para or ortho in respect to the oxygen atom (originally the OH);19 no examples of meta-coupling of alkyl phenols could be found in the literature. In many of the previously reported cases, the formation of CAr–CH2–CAr products was observed for substrates in which CAr–CAr or CAr–O–CAr coupling modes did not lead to stable products. For example, Omura (1984) reported that the decomposition of a C–O-coupling dimer (this coupling product was obtained upon treatment of 2,6-di-tert-butyl-4-methylphenol with I2/KOH in methanol) gave a CAr–CH2–CH2–CAr product (1,2-bis(3,5-di-tert-butyl-4-hydroxyphenyl)ethane).20 Similarly, 2,6-diiodo-4-methylphenol undergoes lateral benzylic oxidation, but at the para methyl group.20 The formation of 17, CAr–CH2–CAr meta-coupling product of b2 with the benzyl radical obtained from o,p-diiodothymol, might, at first, appear to be similar (C2, C4 and C6 positions of diiodothymol were already occupied, i.e. unavailable for coupling followed by aromatization). However, the constitution of 16 (monoiodo derivative), due to the existence of a free position for CAr–CAr or CAr–O–CAr coupling, makes this hypothesis unlikely. To determine whether different reaction conditions employed by Omura (1984) favour benzylic oxidation over CAr–CAr and CAr–O–CAr couplings, we repeated the iodination of 10 (the oxidative coupling of thymol) in methanol instead of an aqueous medium. Nonetheless, the change of solvent did not have a significant influence on the reaction outcome: in general, the same products, including 16 and 17, were obtained and in similar relative amounts.
Based on the mechanisms summarized in Fig. 5 and 6 – these are in agreement with the structures of the purified aristol constituents 7, 14–17 and the previous studies regarding oxidative coupling of (alkylated) phenols – it is reasonable to expect additional possible recombinations of several different O- and C-radicals (including benzylic radicals), obtained from thymol and mono/diiodothymol derivatives (b1–b3), that would yield a much larger number (40) of coupling products belonging to several different families (CAr–CAr, CAr–O–CAr, CAr–CH2–CAr; the structures of all possible coupling products are given in Table S2†). The preliminary results of a GC-MS analysis of crude aristol are in agreement with this. As revealed by a SciFinder search of the CAS database, all of these compounds were unreported, i.e. there were no MS or RI data available in the literature that would allow a straightforward confirmation of their identities.
It is well known that there is a good correlation between gas chromatographic behaviour (retention indices, RI) of a series of (structurally related) compounds and their structural features.21 For example, QSPR (quantitative structure–properties relationship) modelling of RIs of alkyl (poly)sulfides, using simple and easily available 0D and 1D descriptors, enabled the identification of a large number of (new) compounds present in Allium ursinum L. essential oil.22 Although, it is not possible to determine the structure of a compound only from the value of its RI, these retention information, combined with spectral data (MS, for example), might be sufficient to resolve situations when one has several possibilities to choose from. This was precisely the case with the herein studied coupling derivatives of (iodinated) thymol from inseparable fractions of aristol. MS data (m/z corresponding to the molecular ion; this was also the base-peak) were used to divide the detected aristol constituents into appropriate groups of regioisomers (e.g. monomeric or dimeric products; mono- or diiodo-derivatives, etc.). Among the possible regioisomers, their corresponding retention indices could provide an additional criterion that would enable their discernment. We assumed that, if we developed an appropriate QSPR model, we might be able to predict the retention indices for all of the possible regioisomers from any of the coupling families.23 A matching of the calculated (from the QSPR model) and experimental RIs might help us to elucidate the structure of additional aristol constituents, or could at least narrow down the number of potential structures.
Compounds 7, 10–17, with unequivocally determined structures (NMR, IR, HRMS), were the starting point of our QSPR study; the number of compounds initially included in the study was limited, nonetheless, we expected that if the model “worked” (high R2 and low root mean square of the errors, RMSE) on such a small dataset, it would give good results when applied to other possible coupling products.
Usually, RI values are found to correlate with (at least some of) the following simple descriptors: calculated boiling points, critical volume, Connolly solvent excluded volume, partition coefficient, Balaban index, polar surface area, molecular topological index, mol refractivity, Wiener index, the total number of C, H, O and I atoms (13 in total; these are easily obtainable from the compound's structure, using ChemBio3D and EPISuite programs and are readily used in QSPR and QSAR, quantitative structure–activity relationship, studies).23–25 For the development of our QSPR model, we applied the leave-one-out cross-validation approach.26 Although the correlation coefficient obtained using the described input dataset was quite high, RMSE was extremely high (RMSE = 270). An average absolute deviation of RI values recorded on different instruments, but on columns of the same polarity and using the same temperature programs, is generally under 10–15 RI units.27 In that sense, a QSPR model able to predicted RI values with a similar precision (RMSE ranging from 5–10 RI units) should be considered as excellent and reliable.
We assumed that the “monomeric” (10–13) and coupled products (7, 14–17) would interact somewhat differently with the GC stationary phase. Thus, a new QSPR model was built from the data regarding the coupled compounds alone. This resulted in a significant improvement of the model, but RMSE was still quite high (R2 = 1, RMSE = 52 RI units). Many of the used descriptors encoded similar information about the molecules of interest (i.e. they were highly mutually correlated) or turned out not to correlate with RIs at all. Thus, in order to further improve the linear correlation, some of the original descriptors were eliminated. The best results (R2 = 0.9997, RMSE = 1.65 RI units) were obtained for a model that used the predicted boiling points (Bp), the molecular topological index (MTI), and the number of iodine atoms per molecule (NI) as the descriptors (the values of these are listed in Table S2†). This model produced the following regression equation:
| RIpred = 116.09 + 4.07Bp + 0.10MTI − 147.55NI |
The difference between the predicted and experimental RI values was around 1 RI unit or less (e.g. compound 14 RIexp = 2892, RIpred = 2890.7; compound 15 RIexp = 2801, RIpred = 2800.8; compound 17 RIexp = 2923, RIpred = 2923.4).
With this encouraging result, we decided to calculate RI values of all possible coupling products using the derived QSPR equation (Table S2†). Afterwards, the predicted values of the retention indices (RIpred) of the possible coupling products were compared to those experimentally (RIexp) observed with an appropriate molecular mass (i.e. m/z value for the molecular ion). All structures for which the difference between RIexp and RIpred (ΔRI) was above 10 RI units were rejected. In the case of 16 detected (GC-MS analysis) aristol constituents, in addition to 7, 14–17, there was a single possible (based on the mechanism and molecular formula) candidate with a good matching of the values of RIpred and RIexp (ΔRI for these compounds ranged from 0 to 6 RI units). These compounds are listed in Table 3. RMSE calculated over the complete set of the identified coupling products (21) was 3.3 RI units, while R2 = 0.998.
| No | Compound structure | Ma [g mol−1] | RIexpb/RIpredc | % | No | Compound structure | M [g mol−1] | RIexp/RIpred | % |
|---|---|---|---|---|---|---|---|---|---|
| a Molar mass.b RIexp-experimental value.c RIpred-predicted value.d d–g QSPR model does not allow discernment between the structures marked with the same letter in superscript. | |||||||||
| 18d | 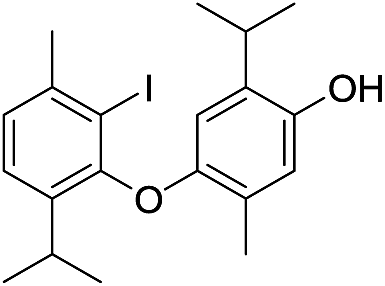 |
424 | 2675/2680 | tr | 31 | 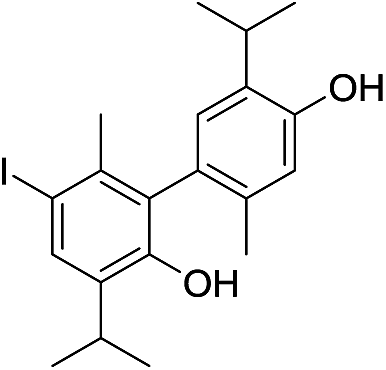 |
424 | 2778/2772 | 0.2 |
| 19 |  |
424 | 2687/2683 | tr | 32 | 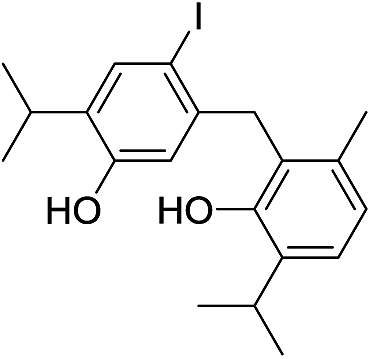 |
424 | 2806/2804 | tr |
| 20 | 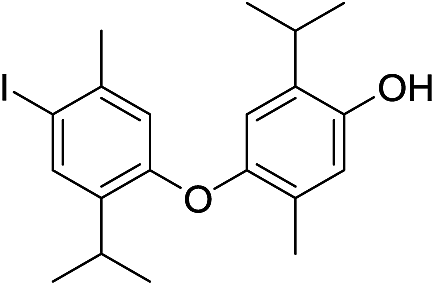 |
424 | 2694/2692 | 0.1 | 33 | 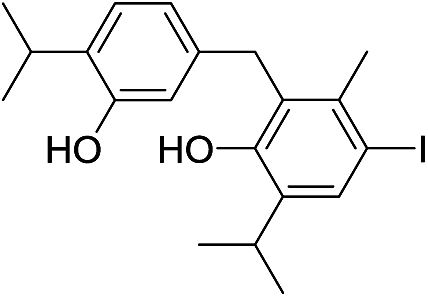 |
424 | 2816/2810 | tr |
| 21 | 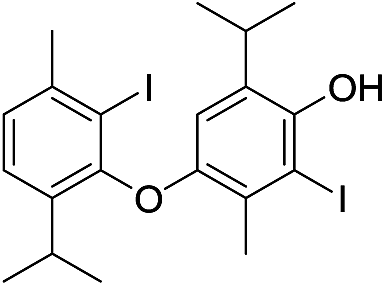 |
550 | 2791/2789 | 2.4 | 34 | 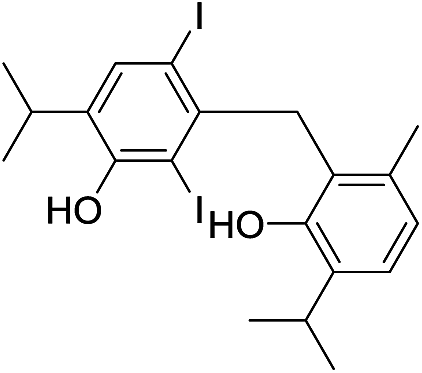 |
550 | 2909/2909 | 0.5 |
| 22 | 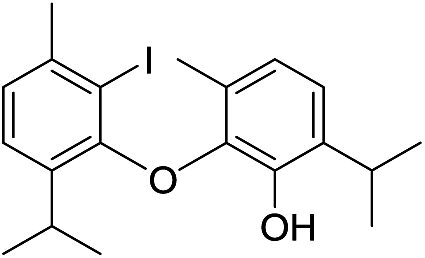 |
424 | 2667/2666 | tr | 36 | 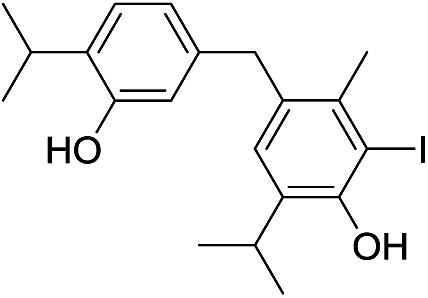 |
424 | 2830/2823 | tr |
| 23 | 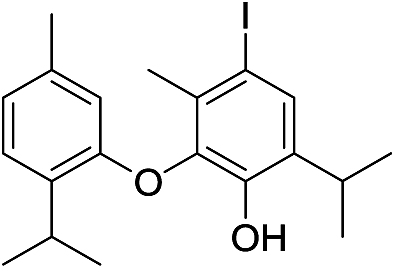 |
424 | 2668/2670 | tr | 37 | 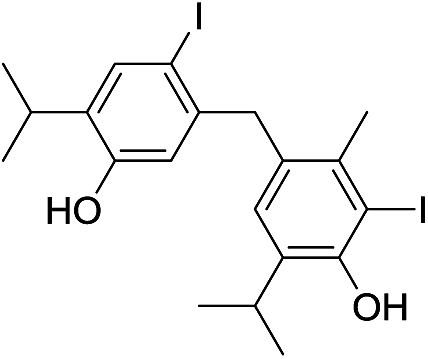 |
550 | 2924/2929 | tr |
| 24d | 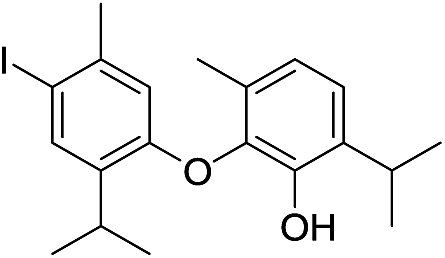 |
424 | 2675/2678 | tr | 38f | 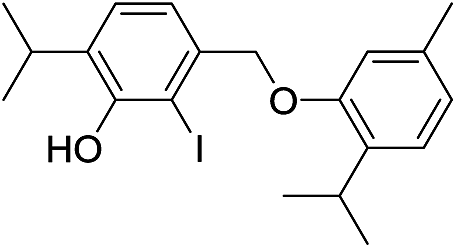 |
424 | 2774/2773 | tr |
| 25 | 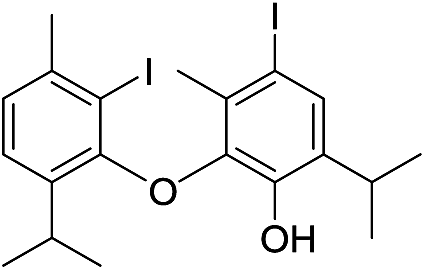 |
550 | 2771/2775 | 1.4 | 39f | 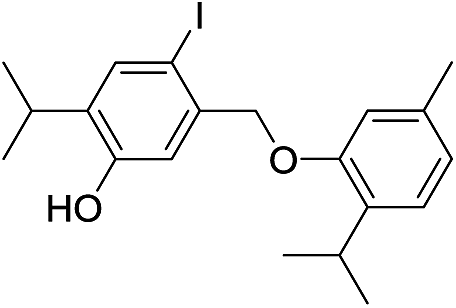 |
424 | 2774/2773 | tr |
| 26e | 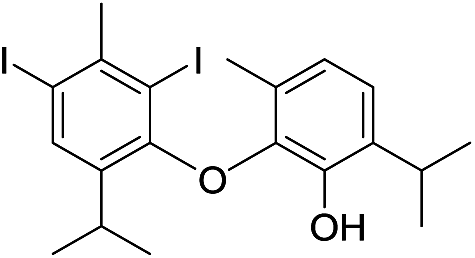 |
550 | 2785/2782 | 4.2 | 40f | 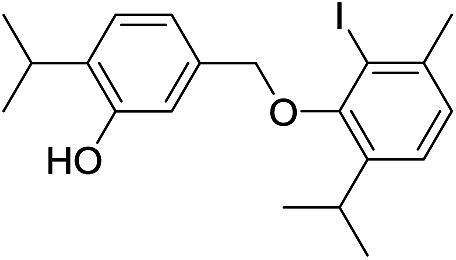 |
424 | 2774/2775 | tr |
| 27e | 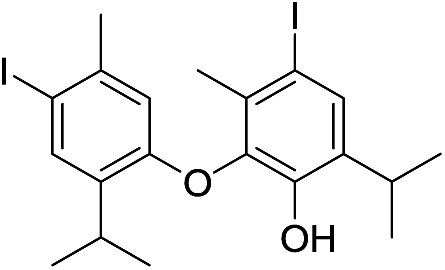 |
550 | 2785/2788 | 4.2 | 41 | 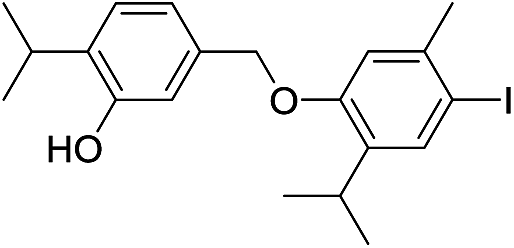 |
424 | 2781/2787 | tr |
| 28 |  |
424 | 2760/2756 | tr | 42 | 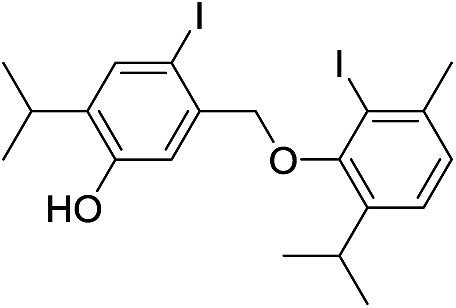 |
550 | 2880/2885 | tr |
| 29 | 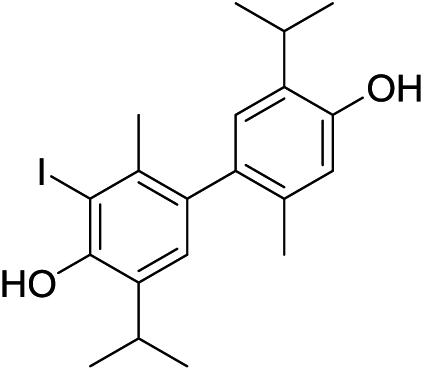 |
424 | 2783/2785 | 4.2 | 43g | 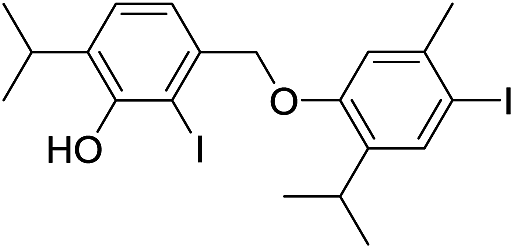 |
550 | 2896/2896 | 1.2 |
| 30f | 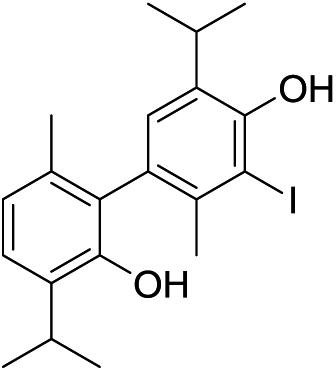 |
424 | 2774/2771 | tr | 44g | 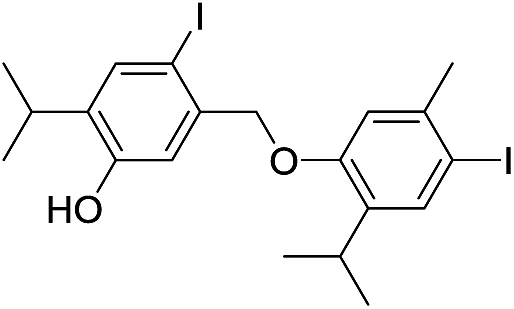 |
550 | 2897/2896 | 1.2 |
In the case of a compound with RIexp = 2675, there were two possible regioisomeric candidates with similar values of their predicted retention indices (entries 18 and 24, Table 3; RIpred = 2680 and 2678, respectively). Similarly, both 26 (RIpred = 2782) and 27 (RIpred = 2788) could represent good matches for the compound with RI = 2785; 30 (RIpred = 2771), 38 (RIpred = 2773), 39 (RIpred = 2773) and 40 (RIpred = 2775) might be the constituent that eluted at RI = 2774. Despite the small differences in ΔRI (these were, except in the case of 18, lower than RMSE of the model), the identity of these aristol constituents cannot be determined on the basis of QSPR alone. The predicted value of the retention indices for structures 43 and 44 (RIpred = 2896 for both) make them excellent candidates for the detected constituents with the experimental RI = 2896 and 2897. However, because of the small differences in the experimental indices, i.e. the same value of the calculated ones, we could not predict which structure corresponded to which of the two detected closely eluting peaks.
Based on the experimental/calculated values of the retention indices of the compounds whose structures are summarized in Table 3, one could draw the following general conclusions: (a) an iodine atom contributed to the value of the retention index with ca. 105 units per I atom; (b) the values of the retention indices were dependent on the coupling mode (for example, CAr–O–CAr compounds had lower indices than their CAr–CAr counterparts); (c) o-iodothymol-derived compounds had the lowest values of RI within a family of compounds with the same coupling mode (the same trend was observed for the order of elution of coupled thymol derivatives from the SiO2 column, i.e., is in accordance with the differences in the polarities of the corresponding compounds, see ESI file†).
Interestingly, a careful inspection of the TIC chromatogram of aristol revealed the presence of 5 additional compounds (1.4% of the total aristol detected peak-areas) with molecular ions at m/z 446 (their corresponding mass spectra were mutually almost identical). The retention indices of these compounds (RI = 2770, 2779, 2906, 2967, 3108) were comparable or significantly (ca. 100 RI units) higher than those of all possible regioisomeric monoiododidehydrodithymols (molecular weight = 424, Fig. S6†). These facts led us to believe that these might be regioisomeric trimers of (non-iodinated) thymol. In addition to these trimers, it seems that, under these conditions, the iodination of thymol yielded several regioisomeric monoiodo trimers (molecular ions at m/z 572; 5.8% of the total TIC peak areas). Unfortunately, the derived QSPR model did not work in the case of these thymol coupling-products (due to the lack of an appropriate learning set), thus, for the time being, their identity remains unresolved.
Conclusions
Despite the fact that the antiseptic drug traded under the name aristol has been on the market for about 130 years and that it has been recommended to be placed on the list of bulk substances allowed for compounding under section 503A of the US Federal Food, Drug, and Cosmetic Act,2 there is a lack of convincing proof in the literature of its currently accepted structure, and the mechanism of its formation is unknown.6–13 Moreover, some of the previously published data seem to be mutually contradictory. To address this problem, we decided to prepare aristol (iodination of thymol, under alkaline conditions)13 and to study its chemical composition, for the first time using modern chromatographic (GC-MS and column chromatography on SiO2) and extensive spectrometric techniques (1D and 2D NMR, FTIR, MS, UV). These clearly showed that aristol does not represent a single compound and that most of the ambiguities concerning the properties of aristol (both chemical and physical) might find their origin in this fact; instead, it is a complex mixture of a large number (25 successfully identified in this work) of structurally closely related products, mainly dimeric coupling products of (iodinated) thymol. We managed to obtain 9 of these in pure state and fully spectrally characterised them. Extensive NMR (1D: 1H and 13C NMR, and selective 1H homodecoupling experiments; 2D: HSQC, HMBC, 1H–1H-COSY and NOESY experiments), HRMS, FTIR and UV analyses, in combination with chemical transformations and quantum mechanical calculation of NMR shifts and coupling constants provided unequivocal proof of the structures of 5 oxidative coupling products (novel compounds: 7, 14–17; Fig. 2), diiodothymol (13), two monoiodothymol regioisomers (11, 12) and unreacted thymol (10). As evident from the structures of the isolated compounds, three different coupling modes were operational: CAr–O–CAr, CAr–CH2–CAr and direct CAr–CAr coupling. The occurrence of CAr–CH2–CAr-type compounds was quite surprising as these were products of an oxidative lateral meta-coupling (with respect to the phenoxy-radical); the later has not been previously reported for any phenol. The mechanism of formation of CAr–CH2–CAr thymol appears to include benzyl radicals that were unknown to form under these conditions.Due to the complexity of the matrix and similarities in their chromatographic behaviour (column chromatography), other aristol constituents (also new compounds; no MS or RI data available) could not be obtained in pure state. Their identification was based on the logic derived from the likely mechanism of oxidative coupling, combined with the prediction of gas chromatographic retention indices. The modelling of retention indices was performed using a QSPR model derived for this very purpose (the isolated compounds served as the training set; readily available structure-derived descriptors were used). Possible hit structures that arose from all possible recombination routes of several different O- and C-radicals (including benzylic radicals) were further filtered using the predicted RIs as the criterion. This approach enabled us to identify 16 additional aristol constituents.
Having in mind the complexity of aristol (a mixture of several dozens of compounds), it is not surprising that different authors previously suggested different structures for this antiseptic drug: its chemical/physical behaviour cannot be satisfactorily explained by only a single structure. The herein presented results urge a revision of the currently accepted formula (composition) of aristol in both primary and secondary literature. Even more importantly, our chemical analyses should provoke further pharmacological studies that might reveal the active principle(s) and mechanism(s) of the desired action of aristol used for more than a century, and, also motivate the development of new potentially more active aristol-like drugs.
Experimental
General
All chemicals were commercially available and were used as received (Sigma-Aldrich, USA; Merck, Germany; Fluka, Germany), except that the solvents were purified by distillation. Silica gel 60, particle size distribution 40–63 μm, was used for “dry flash” chromatography. Thin layer chromatography (TLC) was performed on aluminium plates precoated with Kieselgel 60 F254, layer thickness 0.2 mm. Visualization was accomplished with UV light (254 nm) and by spraying with 50% (v/v) aqueous H2SO4, followed by 5 min heating at 110 °C. The IR measurements (ATR-attenuated total reflectance) were carried out using a FT-IR instrument model 6700 (Thermo Nicolet, USA). UV spectra (in acetonitrile) were measured using a UV-1800 spectrophotometer (Shimadzu, Japan). Melting points were determined on MPM-HV2 melting point meter (Paul Marienfeld GmbH & Co. KG, Lauda-Königshofen, Germany). Microanalysis of carbon and hydrogen were carried out with a Carlo Erba Elemental Analyzer model 1106 (Carlo Erba Strumentazione, Italy) as previously described in Radulović et al.;28 their results agreed favourably with the calculated values. EI-MS and HRMS analyses were performed using a JEOL MStation JMS-700 mass spectrometer with ionization energy of 70 eV, an ionization trap current of 300 μA and a source temperature of 230 °C.29 The error for each elemental composition data is given in units of amu.GC and GC-MS analyses
GC-MS analyses were performed on a Hewlett-Packard 6890N gas chromatograph equipped with fused silica capillary column DB-5MS (5% phenylmethylsiloxane, 30 m × 0.25 mm, film thickness 0.25 μm, Agilent Technologies, USA) and coupled with a 5975B mass selective detector from the same company. The injector and interface were operated at 250 °C and 320 °C, respectively. Oven temperature was raised from 70 °C to 310 °C at a heating rate of 5 °C min−1 and then isothermally held for 10 min. As a carrier gas, He at 1.0 mL min−1 was used. The samples (1 mL of the corresponding solutions in Et2O (1 mg per 1 mL)) were injected in a pulsed-split mode (the flow was 1.5 mL min−1 for the first 0.5 min and then set to 1.0 mL min−1 throughout the rest of the analysis: split ratio, 40:1). MS conditions: ionization voltage 70 eV, acquisition mass range, 35–650 amu, scan time, 0.32 s. The retention indices were calculated relative to the retention times of C12–C30 n-alkanes on the DB-5MS column.
NMR measurements
1H and 13C NMR spectra were recorded on a Bruker Avance III spectrometer (Bruker, Germany) operating at 400 and 100.6 MHz, respectively. 2D experiments (NOESY and gradient 1H–1H COSY, HSQC, HMBC), as well as DEPT-90, DEPT-135 and selective 1H homonuclear decoupling measurements, were run on same instrument with the built-in Bruker pulse sequences. All NMR spectra were measured at 25 °C in CDCl3 with (CH3)4Si as internal standard. Chemical shifts were reported as (δ) in parts per million (ppm) with respect to (CH3)4Si, and coupling constants J values are given in Hertz. The following abbreviations were used to designate multiplicities: s, singlet; br s, broad singlet; d, doublet; pseudo q, pseudo quartet; dqd, doublet of quartets of doublets.Quantitative nuclear magnetic resonance (qNMR)
Quantitative NMR experiments were performed according to a procedure described in Radulović et al.30 To a weighted amount of aristol, dissolved in deuterated chloroform, a known amount of 1,4-dioxane was added as an internal standard (no changes in the appearance of the spectra were noted after the addition of the standard). 1H NMR spectra with 13C decoupling and a large data set (10 points per Hz digital resolution) were recorded. Signal-to-noise ratio of 1000:1 or higher was obtained for all recordings. Parameters were as follows: number of points in the time domain = 32k, spectral width = 10 ppm, O1 = 6.0 ppm, p1 = 45° 1H transmitter pulse, acquisition time = 5 s and number of scans = 1024. After zero-filling and phase and baseline corrections, integration of signals (3.71 ppm for 1,4-dioxane; a non-overlapping signal in the aromatic region of compounds 7, 10–17) was performed. The ratio of the signal integrals was used to calculate the amount of compounds 7, 10–17 in aristol. The results are expressed as weight percentages.
Synthesis of aristol
Three grams (20 mmol) of thymol were dissolved in a solution of 3.2 g (80 mmol) of sodium hydroxide in 100 mL of water. To this solution, at room temperature and with efficient stirring, a solution of 14 g (55 mmol) of iodine and 10 g (60 mmol) of potassium iodide in 50 mL of water was added until an excess of iodine was present. The reaction mixture was filtered through a Büchner funnel, the precipitate washed several times with water and dried in air in the dark. In this way, 5.6 g of aristol was obtained in accordance with the original procedure.13Chromatographic separation of aristol constituents
Air-dried aristol was initially fractionated by gradient “dry flash” column chromatography (hexane–diethyl ether mixtures of increasing polarity) on silica gel. The fractions were pooled based on TLC (2:1 (v/v) hexane and diethyl ether) and GC-MS and this resulted in pooled fractions I–VI. Fractions IV and V represented pure compounds (17 and 16, respectively). Fraction I (1.05 g) was further subjected to another gradient “dry flash” chromatography yielding 19 subfractions. Pure n-hexane (350 mL) was used as the eluent for the first seven subfractions (1–7), followed by 2% (v/v) diethyl ether in n-hexane (subfractions 8–14, 300 mL), 4% (v/v) diethyl ether in n-hexane (subfractions 15 and 16, 100 mL), 10% (v/v) diethyl ether in n-hexane (subfractions 17 and 18, 100 mL) and finally pure diethyl ether (subfraction 19, 50 mL). The solvent was removed in vacuo and the obtained subfractions submitted to GC-MS analysis. The pooled subfractions 1–4 represented a pure compound, 13, while subfractions 5, 7, 12, 13, 15–16, and 17–18 gave pure samples of compounds 11, 15, 14, 7, 10, and 12, respectively.
Spectral characterization of the isolated compounds
ε 3.60), 217 (4.11) and 198 (4.92); FTIR (neat) νmax/cm−1 3167, 2957, 2926, 2867, 1621, 1585, 1516, 1241, 943, 855, 804, 738; RI (DB-5MS) 1291; EIMS m/z 150 (M+, 29%), 135 (100), 136 (9.7), 117 (7.5), 115 (14.2), 107 (6.7), 105 (4.5), 91 (16.1), 79 (3.9), 77 (6.4). For 1H and 13C NMR spectra, see Table 1.ε 3.21), 232 (3.92) and 205 (4.47); FTIR (neat) νmax/cm−1 3372, 2967, 2869, 1555, 1472, 1381, 1370, 1287, 1232, 1151, 880, 874, 714, 568; RI (DB-5MS) 1529; EIMS m/z 276 (M+, 38.4%), 262 (10.1), 261 (100), 134 (26.6), 133 (4.5), 116 (4.1), 115 (8.3), 105 (5.1), 91 (8.6), 77 (8.3); For 1H and 13C NMR spectra, see Table 1.ε 3.23), 233 (3.93) and 205.5 (4.46); FTIR (neat) νmax/cm−1 3382, 2962, 2874, 1543, 1463, 1386, 1377, 1295, 1228, 1157, 886, 880, 707, 570; RI (DB-5MS) 1692; EIMS m/z 276 (M+, 79.8%), 262 (9.9), 261 (100), 134 (39.6), 133 (6.1), 116 (7.0), 115 (11.8), 91 (9.1), 77 (8.6); For 1H and 13C NMR spectra, see Table 1.ε 3.45) and 214.5 (4.66); FTIR (neat) νmax/cm−1 3462, 2959, 2866, 1633, 1581, 1442, 1470, 1386, 1203, 1165, 882, 631, 590; RI (DB-5MS) 1933; EIMS m/z 402 (M+, 91.2%), 388 (9.8), 387 (100), 260 (27.8), 133 (9.5), 115 (7.9), 105 (9.7), 103 (6.3), 77 (9.8); For 1H and 13C NMR spectra, see Table 1.ε 3.73) and 208 (4.81); FTIR (neat) νmax/cm−1 3473, 2959, 2866, 1728, 1662, 1596, 1458, 1391, 1162, 1117, 1020, 884, 800, 609, 570; RI (DB-5MS) 2892; EIMS m/z 550 (M+, 100%), 536 (15.4), 535 (75.7), 393 (5.6), 380 (7.6), 365 (7.3), 260 (29.2), 196 (6.7), 165 (7.1); For 1H and 13C NMR spectra, see Table 2.ε 3.69) and 208 (4.78); FTIR (neat) νmax/cm−1 3480, 2952, 2875, 1738, 1657, 1590, 1464, 1168, 880, 806, 601, 574; RI (DB-5MS) 2863; EIMS m/z 550 (M+, 100%), 536 (9.8), 535 (52.0), 493 (9.1), 393 (8.1), 366 (9.7), 267 (9.8), 260 (24.4), 165 (7.7); For 1H and 13C NMR spectra, see Table 2.ε 3.61) and 206 (4.72); FTIR (neat) νmax/cm−1 3479, 2959, 2921, 2866, 1548, 1465, 1402, 1336, 1237, 1163, 883, 853, 630; RI (DB-5MS) 2801; EIMS m/z 550 (M+, 100%), 535 (10.0), 393 (22.1), 351 (19.1), 275 (13.5), 274 (18.5), 260 (11.7), 115 (13.3), 91 (10.5). For 1H and 13C NMR spectra, see Table 2.ε 3.82) and 202 (4.71); FTIR (neat) νmax/cm−1 3365, 2958, 2926, 2869, 1599, 1394, 1334, 1159, 1098, 894, 857, 754, 604; RI (DB-5MS) 2818; EIMS m/z 424 (M+, 100%), 410 (14.4), 409 (70.1), 381 (5.1), 267 (17.1), 255 (11.8), 254 (8.9), 239 (14.3), 213 (32.3), 211 (16.4), 197 (22.3), 127 (2.9), 91 (4.4), 77 (3.3), 43 (3.9); For 1H and 13C NMR spectra, see Table 2.ε 4.10) and 203 (4.98); FTIR (neat) νmax/cm−1 3457, 2958, 2926, 2870, 1611, 1391, 1158, 905, 854, 731, 602; RI (DB-5MS) 2923; EIMS m/z 550 (M+, 100%), 535 (46.9), 393 (20.6), 381 (16.4), 380 (14.2), 260 (15.4), 253 (21.3), 212 (13.8), 211 (20.4); For 1H and 13C NMR spectra, see Table 2.Methylation of compounds 7, 10–15
A phenol (7, 10, 11, 12, 13, 14, or 15, 0.1 mmol) was added to a suspension of anhydrous potassium carbonate (30 mg, 0.2 mmol) in DMF (5 mL). Methyl iodide (30 mg, 0.21 mmol) was then added and the solution was stirred at room temperature for 24 h. After that, water was added and the reaction mixture was extracted three times with Et2O. The combined organic extracts were washed excessively with brine, and the DMF-free ether extract was dried over anhydrous MgSO4 and concentrated in vacuo. The crude methylation products were directly analysed by GC-MS.32Spectral characterization of the methylated compounds:
Oxidative coupling20 of thymol
A solution of iodine (1.78 g, 7.0 mmol) in methanol was dropwise added to a stirred solution of thymol (1 g, 6.7 mmol) in methanol (20 mL) containing KOH (20 mmol). The mixture was further stirred for 10 min, and afterwards concentrated in vacuo. The reaction mixture was poured into water, neutralized with aqueous HCl (1:1, v/v) and extracted with diethyl ether. The organic layer was washed with aqueous Na2S2O3 solution, dried over anhydrous MgSO4 and the solvent removed on a rotary evaporator. The obtained cure product (2.3 g) was analysed by GC-MS.
Quantitative structure–property modelling (QSPR) of retention indices
Compounds 10–17 were chosen as model compounds. Their experimentally determined RI values (DB-5MS) are given in the subsection of the experimental part-Spectral characterization of the isolated compounds. The retention indices were modelled by taking into account the following calculated/estimated properties/descriptors of 10–17: estimated boiling point, critical volume, Connolly solvent excluded volume, partition coefficient, Balaban index, polar surface area, molecular topological index, molar refractivity, Wiener index, the total number of C, H, O and I atoms. Geometry optimization of compounds 7, 10–17 was performed as described in the following subsection; for all other compounds, MM2 molecular mechanics force field method incorporated in ChemBio 3D Ultra 12.0 software package (CambridgeSoft, USA) was used (MM2 optimisation of 7, 10–17 structures gave geometries equivalent to those based on dispersion-corrected B3LYP-D functional calculations).33 ChemBio 3D Ultra program was used to calculate eight molecular descriptors; only the boiling points were calculated using the EPISuite program.34 The Estimation Programs Interface (EPI) Suite™ predicted different boiling points for isomers 7, 14, 28, 30, 31, while ChemBio 3D gave the same value.The statistical evaluation of the data was performed by the program Microsoft Office Excel 2010.35 Regression analysis was used (confidence interval 95%). To test the quality of the regression equations (given in ESI†), the correlation coefficient (R2), t-value and P-value were utilized as the statistical parameters. The derived QSPR model was validated using the leave-one-out cross-validation approach.26
Computational details
The geometries of all structures of interest were fully optimized without any constraints, with dispersion-corrected B3LYP-D functional (IOp(3/124 = 3)) and ultrafine integration grid. The standard 6-31G(d) basis set was used for H, C and O atoms. Iodine atoms were described with a Stuttgart relativistic ECPs and associated basis set augmented with an additional polarization function (ζd = 0.289) (available from the Basis set Exchange).36–38 The optimized structures were confirmed to be potential energy minima by vibrational frequency calculations at the same level of theory (no imaginary frequencies were found). Additional single-point calculations, based on optimized geometries obtained using the previously described basis set, were performed with a double hybrid mPW2PLYPD functional and a larger basis set (this included the same ECP and basis set for I and all-electron 6-311++G(2d,p) basis set for C, H and O atoms).36 Single point energies were used to compute selected bond dissociation energies (D0), with the approximation that these would be numerically equal to bond dissociation enthalpies (DH298).17 Unrestricted wave functions were used for all of the calculations on radicals. Chemical shifts and coupling constants of the optimized structures of aristol constituents were calculated using GIAO method with B3LYP density functional (6-311++ (d,p) basis set for C, H and O atoms and the same ECP and basis set for I as described above).14,39 All calculations were performed in Gaussian 09.40Acknowledgements
The authors are grateful to the Ministry of Education, Science and Technological Development of the Republic of Serbia (Project 172061) for the financial support of this work. This study is a part of the Ph.D. thesis of Miljana R. Đorđević under the supervision of Niko S. Radulović.References
- J. V. Shoemaker, A Practical Treatise on Materia Medica and Therapeutics: With Especial Reference to the Clinical Application of drugs, F. A. Davis Co, Philadelphia, 1893, vol. II Search PubMed.
- U.S. Food and Drug Administration, Pharmacy Compounding Advisory Committee Meeting, Center for Drug Evaluation and Research, February 23-24, 2015 Search PubMed.
- Department of Health and Human Services-food and drug administration, Fed. Regist., 1999, 64, 996–1003 Search PubMed.
- F. J. S. Gorgas, Dental Medicine: A Manual of Dental Materia Medica and Therapeutics, P. Blakiston Son & Co, Philadelphia, 1895 Search PubMed.
- U. Bengtsson, Dental Materials in Endodontic Therapy, LiTH-IKP-I-137, Linköping, Sweden, 1990 Search PubMed.
- J. Messinger and G. Vortmann, Ber. Dtsch. Chem. Ges., 1889, 22, 2312–2322 CrossRef.
- L. C. Urban, Pharma Rev., 1896, 14, 58–59 CAS.
- P. Dannenberg, Monatshefte für Chemie, 1903, 24, 67–79 CrossRef CAS.
- J. Bougault, J. Pharm. Chim., 1918, 17, 221–227 CAS.
- E. Moles and M. Marquina, An. R. Soc. Esp. Fis. Quim., 1919, 17, 59–83 CAS.
- G. Sanna and T. Zucca, Rend. Semin. Fac. Sci. Univ. Cagliari, 1951, 19, 155–164 CAS.
- The Sigma Aldrich Online Catalog, http://www.sigmaaldrich.com/catalog/product/sigma/t2763?lang=en%26region=SX, accessed May 2016.
- G. H. Woollett, J. Am. Chem. Soc., 1921, 43, 553–561 CrossRef CAS.
- R. Jain, T. Bally and P. R. Rablen, J. Org. Chem., 2009, 74, 4017–4023 CrossRef CAS PubMed.
- D. A. Whiting, in Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming and G. Pattenden, Pergamon, Oxford, 1991, ch. 2.9, pp. 659–703 Search PubMed.
- S. R. Waldvogel and D. Mirk, in Handbook of CH-Transformations, ed. G. Dyker, Wiley-VCH, Weinheim, 2005, ch. 1.4.3, pp. 251–261 Search PubMed.
- S. J. Blanksby and B. G. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- J. Hioe and H. Zipse, in Encyclopedia of Radicals in Chemistry, Biology and Materials, ed. C. Chatgilialoglu and A. Studer, John Wiley & Sons, Inc, Hoboken, New Jersey, 2012, ch. 18, pp. 449–476 Search PubMed.
- W. I. Taylor and A. R. Battersby, Oxidative coupling of phenols, Marcel Dekker, INC, New York, 1967 Search PubMed.
- K. Omura, J. Org. Chem., 1984, 49, 3046–3050 CrossRef CAS.
- B. Škrbić and A. Onjia, Match Communications in Mathematical and in Computer Chemistry, 2006, 55, 287–304 Search PubMed.
- N. S. Radulović, A. B. Miltojević, M. B. Stojković and P. D. Blagojević, Food Res. Int., 2015, 78, 1–10 CrossRef.
- A. R. Katritzky, K. Chen, U. Maran and D. A. Carlson, Anal. Chem., 2000, 72, 101–109 CrossRef CAS PubMed.
- I. G. Zenkevich and A. N. Marinichev, J. Struct. Chem., 2001, 42, 747–754 CrossRef CAS.
- K. L. Goodner, LWT--Food Sci. Technol., 2008, 41, 951–958 CrossRef CAS.
- C. Rojas, P. R. Duchowicz, P. Tripaldi and R. Pis Diez, J. Chromatogr. A, 2015, 1422, 277–288 CrossRef CAS PubMed.
- P. J. Linstrom and W. G. Mallard, NIST Standard Reference Database Number 69, ed. NIST Chemistry WebBook, National Institute of Standards and Technology, Gaithersburg MD, retrieved May 1, 2016, p. 20899, http://webbook.nist.gov Search PubMed.
- N. S. Radulović, M. Z. Mladenović, P. D. Blagojević, Z. Z. Stojanović-Radić, T. Ilic-Tomic, L. Senerovic and J. Nikodinovic-Runic, Food Chem. Toxicol., 2013, 62, 554–565 CrossRef PubMed.
- N. Radulović, Z. Stojanović-Radić, P. Stojanović, N. Stojanović, V. Dekić and B. Dekić, J. Serb. Chem. Soc., 2015, 80, 315–327 CrossRef.
- N. S. Radulović, D. B. Zlatković, T. Ilić-Tomić, L. Senerović and J. Nikodinović-Runić, J. Ethnopharmacol., 2014, 153, 125–132 CrossRef PubMed.
- S. Mishra and A. Singh, Transition Met. Chem., 2005, 30, 163–169 CrossRef CAS.
- R. Leardini, H. McNab, M. Minozzi, D. Nanni, D. Reed and A. G. Wright, J. Chem. Soc., Perkin Trans. 1, 2001, 20, 2704–2710 RSC.
- ChemBio 3D Ultra 12.0, CambridgeSoft, USA, 2010, http://www.cambridgesoft.com/.
- US EPA, Estimation Programs Interface Suite™ for Microsoft® Windows, v. 4.11 [or insert version used] 20, United States Environmental Protection Agency, Washington, DC, USA, 2016 Search PubMed.
- Microsoft Office Excel, 2010, http://www.microsoft.com/.
- A. I. Konovalov, A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2014, 136, 13410–13425 CrossRef CAS PubMed.
- D. Feller, J. Comput. Chem., 1996, 17, 1571–1586 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
- P. R. Rablen, S. A. Pearlman and J. Finkbiner, J. Phys. Chem. A, 1999, 103, 7357–7363 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc, Wallingford CT, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11296j |
| This journal is © The Royal Society of Chemistry 2016 |