
Identification of new p300 histone acetyltransferase inhibitors from natural products by a customized virtual screening method†
Guo-Bo Lia,
Lu-Yi Huanga,
Hui Lia,
Sen Jia,
Lin-Li Lib and
Sheng-Yong Yang*a
aState Key Laboratory of Biotherapy/Collaborative Innovation Center for Biotherapy, West China Hospital, West China Medical School, Sichuan University, Sichuan 610041, China. E-mail: yangsy@scu.edu.cn; Fax: +86-28-85164060; Tel: +86-28-85164063
bKey Laboratory of Drug Targeting and Drug Delivery System of Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu, Sichuan 610041, China
First published on 20th June 2016
Abstract
Herein, we sought to discover new p300 HAT inhibitors from natural products by a customized structure-based virtual screening method. The natural compounds NP-2 (spinosine), NP-3 (palmatine), NP-9 (venenatine), and NP-15 (taxodione) were found to be potent p300 HAT inhibitors, of which IC50 values are 0.69 μM, 1.05 μM, 0.58 μM, 4.85 μM, respectively.
p300 (also called EP300 or KAT3B) and its CBP paralog (CREBBP or KAT3A) are crucial coactivators for gene regulation existing in humans and most higher eukaryotes, which contain histone acetyltransferase (HAT) activity.1 p300 is a large protein of ∼300 kDa and contains several well-defined domains, including a core catalytic HAT domain and a bromodomain (BRD) that recognizes N-acetylated substrates.1 In addition to histones, the p300 HAT domain has been observed to acetylate more than 100 other substrate proteins, including p53, NF-κB, STAT3, and HSP90.1–3 p300 has been reported to be involved in many important signalling pathways such as the cAMP, Notch signalling, estrogen, stress response, and DNA damage response pathway, and regulates multiple cellular processes for example cell proliferation, differentiation, apoptosis, and DNA repair.1,4,5 Aberrant p300 HAT activity is thought to be a crucial factor contributing to several human diseases, including cancer, inflammation, cardiac disease, and Hungtington's disease.1,6 Therefore, the development of p300 HAT inhibitors would be an impetus to probe related molecular mechanisms and to develop potential new agents for the treatment of associated diseases.
To date, many p300 HAT inhibitors have been reported (Fig. S1†). The first potent p300 HAT inhibitors were bi-substrate analogues such as Lys-CoA (Ki of 19 nM), which proved a promising tool for studying p300 HAT mechanism by crystallography and kinetics, but the inherent complexity of these compounds is limiting for pharmacological applications. Shortly thereafter, high-throughput screening experiments led to a number of small-molecule synthetic agents such as windorphen and L001, and natural products such as anacardic acid, garcinol, plumgin, quercetin, and curcumin as p300 HAT inhibitors (Fig. S1†), and almost all of them only have moderate inhibitory activities. Recently, virtual screening approaches were applied and resulted in some new potent inhibitors for instance C646 and Cpd17 (Fig. S1†). C646 is fairly potent against p300 (Ki of 400 nM)7 and was proven to be an attractive probe in molecular mechanism studies while lacking pharmacologic liabilities commonly associated with the 3-methyl-4-methylene-1H-pyrazol-5(4H)-one scaffold that may potentially cause off-target effects.8 Taken together, there remains a current lack of highly potent and selective p300 HAT inhibitors available.
Through analysis of the structures of p300 HAT in complex with acetyl coenzyme A (acetyl-CoA) substrate or analogues,2,9 we observed that the acetyl-CoA binding site is a long and narrow-shaped channel, and the phosphate motif of acetyl-CoA forms multiple hydrogen-bonding and electrostatic interactions with the key residues Arg1410, Thr1411, and Trp1466, which probably are the most important factor for acetyl-CoA binding (Fig. S2†). We thus hypothesized that compounds with a complementary long and narrow shape capable of forming hydrogen-bonding interactions with these three key residues are likely to have p300 HAT inhibition potential. Natural products are a rich source of such compounds, because natural products have a high degree of stereochemistry and a wide range of pharmacophore features,10 potentially allowing them to fit in the p300 HAT binding site. In addition, natural products have another advantage over synthetic compounds of having the ‘metabolite-likeness’ property, which contributes to their ability to be biologically active and biocompatible, an essential prerequisite for pharmacological applications.10 Based on the key features of the p300 HAT binding site and the unique advantages of natural products, in this study we sought to identify new p300 HAT inhibitors from natural product collections using a customized structure-based virtual screening method.
The virtual screening protocol used here is graphically shown in Fig. 1. Details of this protocol can be found in ESI.† In brief, a structural library of 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 969 natural products was assembled from four commercially available natural product collections. These natural products were then filtered by three rules including (i) molecule weight ≤ 500 Da , (ii) number of polar atoms ≤ 10, and (iii) number of rotatable bonds ≤ 10, with the aim of excluding the large complicated natural products, which are not easy to screen by molecular docking. This step had 21622 compounds pass through. Then, the resulting compounds were subjected to molecular docking analysis using the Autodock VINA docking program11 to predict binding modes and affinities. A total of 17653 compounds with VINA score lower than −8.0 kcal mol−1 were identified and subsequently submitted to hydrogen-bonding analysis using an in-house program derived from ID-score method,12 which was designed to select compounds that are likely to form hydrogen-bonds with each of the three key residues (Arg1410, Thr1411, and Trp1466). The hydrogen-bonding analysis resulted in 1595 compounds. These compounds were further inspected visually to check whether the predicted binding poses are reasonable and to select diverse natural compounds as potential p300 inhibitors. To this end, a list of 110 natural compounds (see ESI Table S1†) were obtained, which were expected to provide potential new hits as well as information for identifying p300 HAT inhibitors. From these compounds, we selected fifteen commercially available compounds (Table 1) for testing against p300 HAT activity in vitro.
969 natural products was assembled from four commercially available natural product collections. These natural products were then filtered by three rules including (i) molecule weight ≤ 500 Da , (ii) number of polar atoms ≤ 10, and (iii) number of rotatable bonds ≤ 10, with the aim of excluding the large complicated natural products, which are not easy to screen by molecular docking. This step had 21622 compounds pass through. Then, the resulting compounds were subjected to molecular docking analysis using the Autodock VINA docking program11 to predict binding modes and affinities. A total of 17653 compounds with VINA score lower than −8.0 kcal mol−1 were identified and subsequently submitted to hydrogen-bonding analysis using an in-house program derived from ID-score method,12 which was designed to select compounds that are likely to form hydrogen-bonds with each of the three key residues (Arg1410, Thr1411, and Trp1466). The hydrogen-bonding analysis resulted in 1595 compounds. These compounds were further inspected visually to check whether the predicted binding poses are reasonable and to select diverse natural compounds as potential p300 inhibitors. To this end, a list of 110 natural compounds (see ESI Table S1†) were obtained, which were expected to provide potential new hits as well as information for identifying p300 HAT inhibitors. From these compounds, we selected fifteen commercially available compounds (Table 1) for testing against p300 HAT activity in vitro.
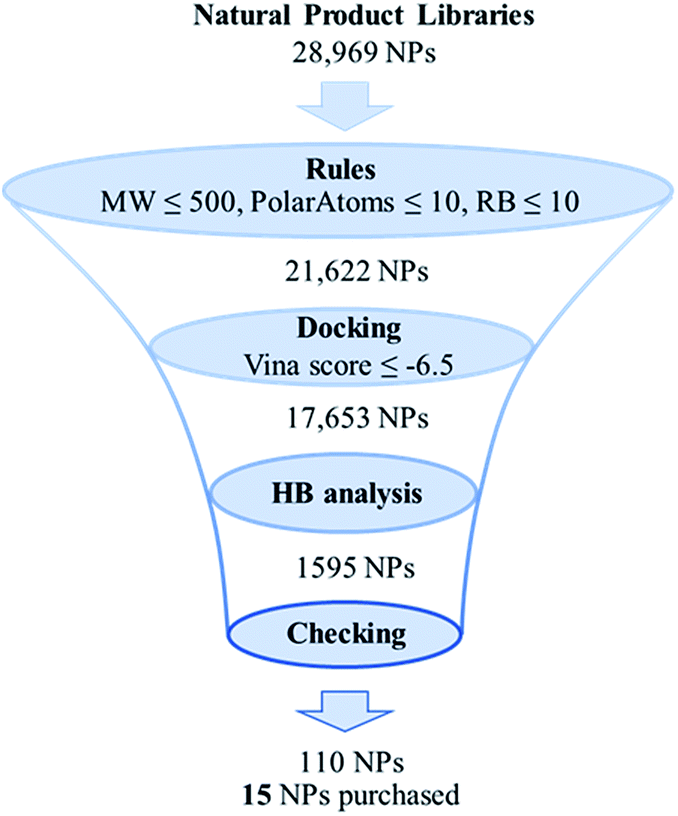 | ||
| Fig. 1 The proposed virtual screening protocol for identifying new p300 HAT inhibitors from natural products. | ||

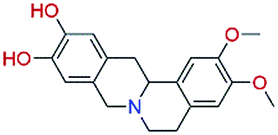






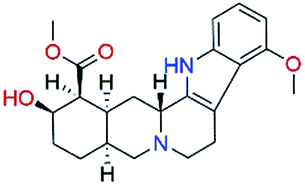





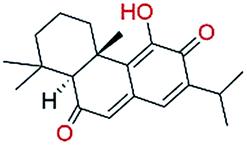
The in vitro assays were carried out using an isotope labelling method as described in ESI† Experimental methods. The compounds were first tested at a fixed concentration of 10 μM (Table 1). We observed 10 compounds with more than 15% inhibitory activity against p300 HAT at the concentration of 10 μM, and 4 compounds including NP-2, NP-3, NP-9, and NP-15 with more than 50% inhibition. We subsequently measured the IC50 values for NP-2, NP-3, NP-9, and NP-15 as well as the well-known p300 HAT inhibitor C646 for comparison. These compounds exhibited good dose–response relationships (Fig. 2), and under the assay conditions used, the IC50 values for NP-2, NP-3, NP-9, and NP-15 were determined to be 0.69 μM, 1.05 μM, 0.58 μM, 4.85 μM, respectively. Of the 4 compounds, NP-2, NP-3, and NP-9 show more potent p300 HAT inhibitory activities than C646 (IC50 of 1.83 μM).
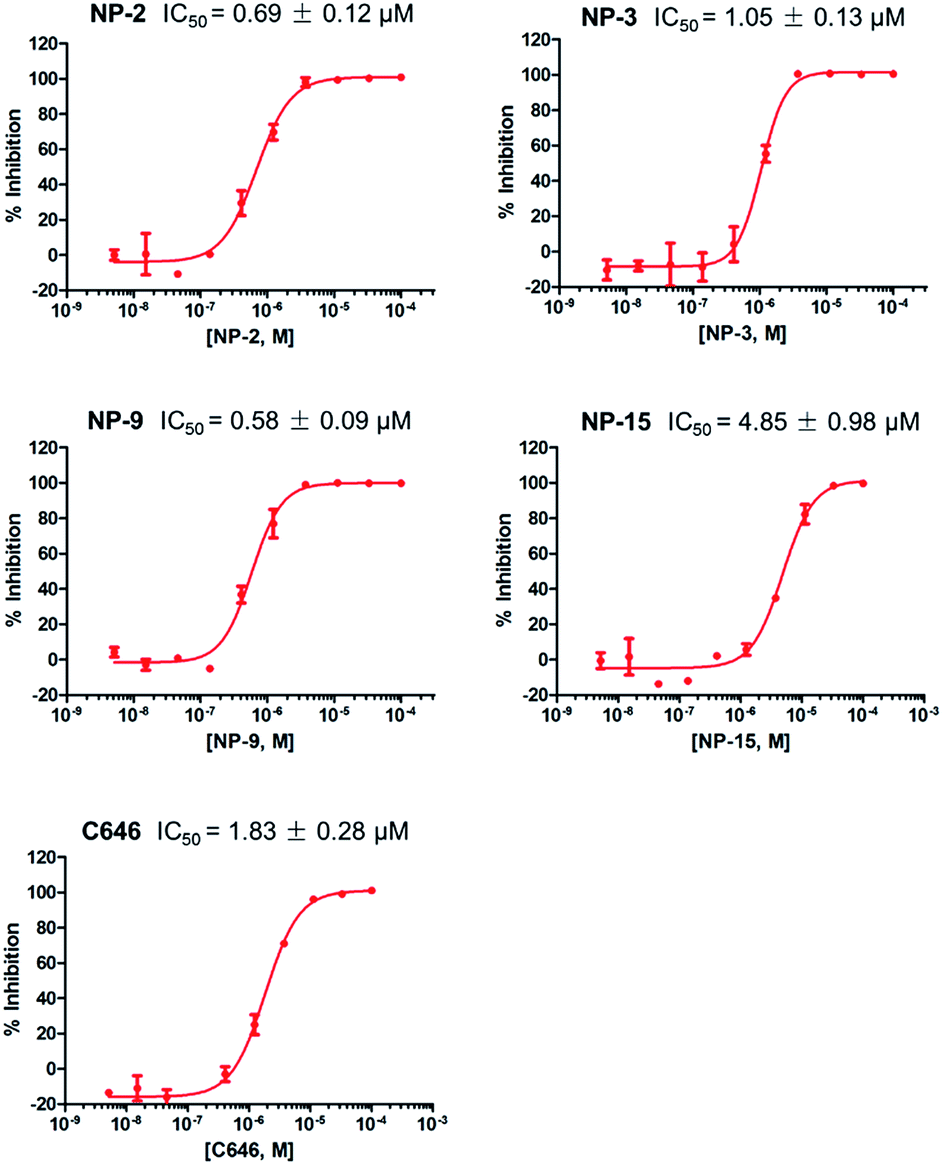 | ||
| Fig. 2 The IC50 curves of compounds NP-2, NP-3, NP-9, NP-15, and C646. | ||
Until this study, none of the compounds NP-2, NP-3, NP-9, and NP-15 nor their scaffolds have been reported as p300 HAT inhibitors. Compound NP-2 (spinosine) is a berbine alkaloid, which can be isolated from the trunk bark and roots of Annona spinescens.13 The compound NP-2 was tested as a racemic mixture. Fig. 3A shows the predicted binding modes of NP-2(R) and NP-2(S) to be very similar. Most importantly, both stereoisomers binding modes have hydrogen-bonding interactions with the three key residues (ARG1410, THR1411, and TYR1467), suggesting that both may have inhibitory activities against p300 HAT. The compound NP-3 (palmatine) is a protoberberine alkaloid,14 which has a similar chemical scaffold to that of NP-2, and results in similar predicted binding modes showing hydrogen-bonding interactions with the three key residues (Fig. 3B). The most active compound NP-9 (venenatine) is an indole alkaloid,15 and has been reported to have good antifungal activity. NP-9 is also predicted to have a similar binding mode to NP-2 and NP-3. Another common feature of these three alkaloid compounds is that they have a positively charged center, which seems to form ionic interactions with ASP1399 (Fig. 3A–C). Compound NP-15 (taxodione) is an abietane diterpenoid, which can be separated from the roots of a Kenyan medicinal plant, Clerodendrum eriophyllum.16 NP-15 has a simple, different scaffold compared with NP-2, NP-3 and NP-9, and its p300 HAT inhibitory activity may be due to predicted hydrogen bonds with the three key residues observed in docking studies (Fig. 3D). Through overlapping the predicted binding modes, NP-2, NP-3, NP-9, and NP-15 were observed to have similar binding modes with the p300 HAT substrate analogue Lys-CoA (Fig. 3E and F). These results suggest that the hydrogen-bonding interactions with the key residues Arg1410, Thr1411, and Trp1466 might be essential pharmacophore features of p300 HAT substrate and inhibitor binding, and the virtual screening method presented here is an effective, feasible method for identifying novel p300 HAT inhibitors.
 | ||
| Fig. 3 The predicted binding modes of NP-2 (A), NP-3 (B), NP-9 (C), and NP-15 (D) within p300 HAT domain, and the co-crystal p300 inhibitor Lys-CoA (E) as well as overlapping with the predicted binding modes (F). | ||
We subsequently tested the inhibitory activities of NP-2, NP-3, NP-9 and NP-15 against the other three histone acetyltransferase including PCAF, HAT1, and KAT7 to investigate their selectivity profiles. As shown in Table 2, NP-2, NP-3, NP-9 and NP-15 exhibited moderate activity against PCAF (19%, 53%, 48% and 63% inhibitory activities at concentration of 10 μM, respectively), and all these compounds displayed no or very weak inhibitory activity against HAT1 and KAT7. The inhibitory potencies (IC50) of NP-2, NP-3, NP-9, and NP-15 against PCAF were 14.13 μM, 10.0 μM, 27.1 μM, 7.16 μM, respectively (for comparison, the PCAF inhibitor garcinol is 20.7 μM, see ESI Fig. S3†), which are lower than that against p300. Bioinformatics analyses revealed that the active site of p300 HAT is significantly different from that of PCAF, HAT1, and KAT7 both in sequence and protein tertiary structure, and the p300 key residues Arg1410, Thr1411, and Trp1466 are not conserved (see ESI Fig. S4 and S5†), which may explain why these natural compounds possess considerable selectivity for p300. We further determined the inhibitory activity of NP-2, NP-3, NP-9 and NP-15 against several human cancer cell lines by cell viability assays as described in ESI† Experimental methods. NP-2, NP-3, and NP-9 showed weak inhibitory activities against MDA-MB-231, ZR-75-1, LNCaP, DU 145, PANC-1, HCT 116, DLD-1, and SH-SY5Y cancer cell lines (Table S2†), reflecting that these cancer cell lines may not be sensitive to selective p300 inhibitors, and implying that these natural products are non-cytotoxic compounds. In contract, NP-15 displayed submicromolar inhibitory activity against almost all the tested cancer cell lines (Table S2†), probably due to off-target effects. Taken together, these natural products should be useful in further efforts to develop new p300 HAT inhibitors for the treatment of related diseases.
| Cpd ID | Inhibition%@10 μM | ||
|---|---|---|---|
| PCAF | HAT1 | KAT7 | |
| NP-2 | 19 | −23 | −17 |
| NP-3 | 53 | 7 | 8 |
| NP-9 | 48 | −2 | −2 |
| NP-15 | 63 | −5 | 5 |
Conclusions
In summary, we present a customized structure-based virtual screening method for identifying new p300 HAT inhibitors from natural products based on the hypothesis that hydrogen-bonding interactions with three key residues Arg1410, Thr1411, and Trp1466 are essential pharmacophore features for p300 HAT inhibitor binding. A total of 110 natural products were selected as potential p300 HAT inhibitors by this method, from which 15 compounds were purchased and screened by in vitro assays. The natural compounds NP-2 (spinosine), NP-3 (palmatine), NP-9 (venenatine), and NP-15 (taxodione) were found to have good inhibitory activities against p300 HAT, of which IC50 values are 0.69 μM, 1.05 μM, 0.58 μM, 4.85 μM, respectively. These results indicate the customized method is an effective, feasible strategy to identify potent p300 HAT inhibitors. Collectively, this study is expected to provide a useful virtual screening strategy and new hit/lead compounds for identifying more potent and selective p300 HAT inhibitors for probing cellular molecular mechanisms as well as for drug discovery & development.Acknowledgements
We are very grateful to Michael A. McDonough for revising the manuscript. This work was supported by the 973 Program (2013CB967204), the National Natural Science Foundation of China (81502989, 81473140, 815733499), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT13031).Notes and references
- B. M. Dancy and P. A. Cole, Chem. Rev., 2015, 115, 2419–2452 CrossRef CAS PubMed.
- J. Maksimoska, D. Segura-Pena, P. A. Cole and R. Marmorstein, Biochemistry, 2014, 53, 3415–3422 CrossRef CAS PubMed.
- Y. Yang, R. Rao, J. Shen, Y. Tang, W. Fiskus, J. Nechtman, P. Atadja and K. Bhalla, Cancer Res., 2008, 68, 4833–4842 CrossRef CAS PubMed.
- M. Breaux, K. Lewis, L. Valanejad, P. Iakova, F. Chen, Q. X. Mo, E. Medrano, L. Timchenko and N. Timchenko, Mol. Cell. Biol., 2015, 35, 3005–3016 CrossRef CAS PubMed.
- M. Hirota, K. Watanabe, S. Hamada, Y. P. Sun, L. Strizzi, M. Mancino, T. Nagaoka, M. Gonzales, M. Seno, C. Bianco and D. S. Salomon, Cell. Signal., 2008, 20, 1632–1641 CrossRef CAS PubMed.
- Y. Liu, L. Wang, J. Predina, R. Han, U. H. Beier, L.-C. S. Wang, V. Kapoor, T. R. Bhatti, T. Akimova, S. Singhal, P. K. Brindle, P. A. Cole, S. M. Albelda and W. W. Hancock, Nat. Med., 2013, 19, 1173–1177 CrossRef CAS PubMed.
- E. M. Bowers, G. Yan, C. Mukherjee, A. Orry, L. Wang, M. A. Holbert, N. T. Crump, C. A. Hazzalin, G. Liszczak, H. Yuan, C. Larocca, S. A. Saldanha, R. Abagyan, Y. Sun, D. J. Meyers, R. Marmorstein, L. C. Mahadevan, R. M. Alani and P. A. Cole, Chem. Biol., 2010, 17, 471–482 CrossRef CAS PubMed.
- J. B. Baell, Future Med. Chem., 2010, 2, 1529–1546 CrossRef CAS PubMed.
- X. Liu, L. Wang, K. Zhao, P. R. Thompson, Y. Hwang, R. Marmorstein and P. A. Cole, Nature, 2008, 451, 846–850 CrossRef CAS PubMed.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- G.-B. Li, L.-L. Yang, W.-J. Wang, L.-L. Li and S.-Y. Yang, J. Chem. Inf. Model., 2013, 53, 592–600 CrossRef CAS PubMed.
- E. F. Queiroz, F. Roblot, A. Cavé, M. de, Q. Paulo and A. Fournet, J. Nat. Prod., 1996, 59, 438–440 CrossRef CAS PubMed.
- R. Marek, P. Sečkárová, D. Hulová, J. Marek, J. Dostál and V. Sklenár, J. Nat. Prod., 2003, 66, 481–486 CrossRef CAS PubMed.
- U. Singh, B. Sarma, P. Mishra and A. Ray, Folia Microbiol., 2000, 45, 173–176 CrossRef CAS.
- F. Machumi, V. Samoylenko, A. Yenesew, S. Derese, J. O. Midiwo, F. T. Wiggers, M. R. Jacob, B. L. Tekwani, S. I. Khan and L. A. Walker, Nat. Prod. Commun., 2010, 5, 853 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11240d |
| This journal is © The Royal Society of Chemistry 2016 |