
Self-healing of thermally-induced, biocompatible and biodegradable protein hydrogel†
Jun Chena,
Xiaoyu Maa,
Qiuchen Donga,
Donghui Songb,
Derek Hargroveb,
Sahil R. Vorac,
Anson W. K. Macd,
Xiuling Lub and
Yu Lei*ac
aDepartment of Biomedical Engineering, University of Connecticut, Storrs, CT 06269, USA. E-mail: ylei@engr.uconn.edu; Fax: +1-860-486-2959; Tel: +1-860-486-4554
bDepartment of Pharmaceutical Sciences, University of Connecticut, Storrs, CT 06269, USA
cDepartment of Chemical and Biomolecular Engineering, University of Connecticut, Storrs, CT 06269, USA
dPolymer Program, Institute of Materials Science, University of Connecticut, Storrs, CT 06269, USA
First published on 7th June 2016
Abstract
Serum albumin is the most abundant protein in the circulatory system to transport fatty acids, metabolites and drugs. In this study, a highly biocompatible protein hydrogel was prepared from bovine serum albumin (BSA) via thermal treatment. A circular dichroism study indicates that thermally-induced partial unfolding of the protein molecules exposes the buried hydrophobic groups in the core to the environment, thus leading to the formation of fine stranded 3-D networks. By controlling the heating temperature and protein concentration, the mechanical strength and structural stability of the as-prepared BSA hydrogel can be facilely manipulated. The moderate denaturation of the protein within the hydrogel system allows repetitive self-healing after damage when moderate heat was induced. The tensile strength and break strain of fully healed protein hydrogel were recovered to almost 100% of the original strength and elongation abilities. Additionally, the good biocompatibility of this hydrogel system was demonstrated through in vitro cytotoxicity analysis first. Furthermore, in vivo experiments using immunocompetent mice show that the subcutaneously injected hydrogel in mice can be fully degraded with negligible acute inflammatory response, indicating excellent in vivo biocompatibility. These features indicate that the as-developed self-healing protein hydrogel system with good biocompatibility and biodegradability holds great potential in the field of biomedical engineering.
1. Introduction
Hydrogels are three-dimensional networks of hydrophilic natural or synthetic polymers.1–4 Due to their high water content, tissue-mimic physical and mechanical properties and biocompatibility, hydrogels have the potency to be used in numerous biomedical applications such as tissue repair, reconstructive surgery, drug delivery and cell encapsulation.5,6 Over the past decades, hydrogels have been extensively used in drug delivery and tissue engineering due to their multiple advantageous characteristics.7–14 In addition, biodegradable hydrogels are specifically attractive for tissue engineering. Biodegradable materials can provide supports during the healing process with a degradation rate matching the rate of new tissue formation.15–17 Both synthetic and natural biodegradable hydrogels have been designed as cell and/or growth factor carriers for bone and cartilage regeneration and orthopedic tissue engineering to improve defect healing and reduce secondary site morbidity simultaneously.15–17 Therefore, hydrogels with good biocompatibility and biodegradability are highly demanded for a range of biomedical applications.On the other hand, in most biological systems, damage or failure could be automatically healed without any external stimuli. Inspired by nature, self-healing hydrogels composed of synthetic or natural materials have been witnessing rapid development nowadays. Among various applications of self-healing hydrogels, recent study using ultrasound to mimic sonophoresis process provides a new strategy for controlled drug release in which high-dose bursts of drug could be achieved by exposure of the hydrogel to ultrasound treatment and these bursts could be lowered down to a low baseline via self-healing of the hydrogel system after removal of ultrasound.8 In this way, self-healing hydrogels offer a new strategy to a drug delivery system which can be systematically controlled with both spatial and temporal release.
Up to date, natural biomacromolecules such as collagen, gelatin, chitosan, hyaluronic acid and alginate have all been utilized to fabricate biodegradable hydrogel due to their high similarity to the extracellular matrix.18 However, bovine serum albumin (BSA) is frequently employed as the model protein released from different matrices rather than the building block of the biodegradable networks. Serum albumin is a protein synthetized by the liver to transport fatty acids, metabolites and drugs.19–22 It is the most abundant protein in the circulatory system.23 It has been reported that various hydrogels could be synthesized from BSA via thermal or chemical cross-linking methods.24 To differentiate from previous work, herein a thermal-induced BSA hydrogel system was systematically investigated to elucidate the gelation conditions and mechanism, and then its self-healing property was validated for the first time by tensile test, followed by the evaluation of its biocompatibility and biodegradability through in vitro and in vivo studies. The as-developed self-healing protein hydrogel system with good biocompatibility and biodegradability holds great potential in the field of biomedical engineering.
2. Experiments
2.1 Chemicals and materials
BSA (≥98%, agarose gel electrophoresis lyophilized, MW = 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 430 Da) and phosphate buffer saline (PBS) were purchased from Sigma-Aldrich (MO, USA). Standard stock solutions of Na2HPO4 and NaH2PO4 (20 mM) were mixed at an appropriate ratio to prepare phosphate buffer solution (PB, 20 mM, pH 7.4). One pouch of PBS pellet was dissolved in 1 l deionized water to yield 0.01 M phosphate buffered saline with 0.138 M NaCl and 0.0027 M KCl at pH 7.4. All aqueous solutions were prepared with deionized water (18.2 MΩ cm) from a Millipore water purification system.
430 Da) and phosphate buffer saline (PBS) were purchased from Sigma-Aldrich (MO, USA). Standard stock solutions of Na2HPO4 and NaH2PO4 (20 mM) were mixed at an appropriate ratio to prepare phosphate buffer solution (PB, 20 mM, pH 7.4). One pouch of PBS pellet was dissolved in 1 l deionized water to yield 0.01 M phosphate buffered saline with 0.138 M NaCl and 0.0027 M KCl at pH 7.4. All aqueous solutions were prepared with deionized water (18.2 MΩ cm) from a Millipore water purification system.
2.2 Preparation of BSA hydrogel
BSA solutions in different concentrations (5–30%, w/v) were prepared by dissolving different weight of BSA powder into 20 mM pH 7.4 phosphate buffer solution. The sol–gel phase transitions of BSA solution with different BSA concentrations were determined by dynamic rheological analysis.25 Each sample was placed onto the object table and then slowly heated from 20 to 100 °C at temperature intervals of 2 °C (maintained at each temperature for 20 min).2.3 Characterization
Attenuated total reflectance FT-IR spectra were obtained with a Thermo Nicolet IR 560 system (Thermo Electron Corporation, PA) in the range of 4000–800 cm−1. Each spectrum was taken as the average of 128 scans at a resolution of 4 cm−1. All the IR spectra were analyzed using the software of Ominic from Thermo Electron Corporation.Circular dichroism (CD) spectra were acquired using Jasco J-710 CD-spectropolarimeter (Jasco Analytical Instruments, Japan) in the wavelength of 201–250 nm. 1% BSA solution were diluted to 0.003% and then heated at different temperature (20–100 °C) for 20 min to study the conformational changes of BSA during the heating process.
Observations of morphologies and pore structures of the BSA hydrogels in different concentrations were obtained by JEOL 6335 Field Emission Scanning Electron Microscope (FESEM) at an acceleration voltage of 10 kV. All samples were lyophilized to maintain porous structures of the hydrogels. Prior to SEM observations, specimens were cut by a razor and sputtered coated with Au/Pd alloy for better resolution.
Mechanical measurements of the hydrogels were conducted on TA instruments Q800. Compressive tests were performed from a constant preload force of 0.01 N with ramp force of 0.5 N min−1 up to 18 N at 37 °C. Hydrogel samples with 20%, 25% and 30% BSA for compressive tests were prepared by incubating at 80 °C for 20 min. All specimens were prepared in the form of discs with 5 mm in diameter and 3 mm in length. Tensile tests were performed from a constant preload force of 0.01 N with ramp force of 0.05 N min−1 up to 18 N at 37 °C. 30% BSA hydrogels heated at different temperature were cut by two parallel razors into thin films with dimensions as 6 mm length × 4 mm width × 1 mm thickness.
Dynamic rheological measurements of the hydrogels were conducted using an AR-G2 rheometer (TA Instrument) with a 20 mm (dia.) parallel-plate fixture. Small oscillatory shear with a strain amplitude of 1% and a frequency of 6.3 rad s−1 was applied to the sample. The storage modulus (G′) and loss modulus (G′′) were recorded as the temperature increased from 20 °C to 100 °C. At a given temperature, the sample was allowed to equilibrate for 20 minutes before the G′ and G′′ values were taken. Small amount of silicone oil was added to the rim to prevent water evaporation during rheological measurements.
2.4 In vitro compatibility analysis
Human lung carcinoma A549 cells were used for cytotoxicity analysis of the hydrogel. 30% BSA hydrogel in dry weight of 300 mg were soaked in 15 ml (2%, w/v) Dulbecco's Modified Eagle Medium (DMEM) and cultured for 24 hours at 37 °C in the incubator. After that, the 2% hydrogel extract solution with DMEM was diluted to 0.2, 0.4, 0.6, and 1%, respectively. 0.1 ml per well of the 5 × 104 cells per ml suspensions were seeded to the wells of 96-well culture plates and then cultured in the incubator for 24 h in order to make 5000 A549 cells per well attached to the bottom of the culture plate. After the cell medium was removed, 0, 0.2, 0.4, 0.6, 1, and 2% hydrogel extract solution were used to fill the wells (3 wells for each sample) and three wells with DMEM only as the blank. After incubation for 24 h, the hydrogel extract solutions were replaced by the fresh medium. Then, 10 μl of Cell Counting Kit-8 (CCK-8) was added to each well. The absorption of each well at 450 nm was recorded after 1 h incubation by a microplate reader, and the cell survival percentage was calculated according to cell survival (%) = (Asample − Ablank)/(A0 − Ablank) × 100%, where Asample, Ablank, and A0 are the mean values of three parallel absorptions in the extract solutions, blank, and culture medium, respectively.2.5 In vivo compatibility and biodegradation analysis
All studies were performed following the University of Connecticut Institutional Animal Care and Use Committee approved protocol. In this investigation, 30% BSA aqueous solution was incubated at 70 °C for 20 min to form the hydrogel. After sterilization by autoclave, the hydrogel was ready for injection. Two male immunocompetent C57BL/6 mice were anesthetized by isoflurane before injection. Hairs on the implantation areas were removed by depilation and the naked regions were cleaned with alcohol swabs. Then the specimen was subcutaneously injected in two mice using a modified 18-gauge indwelling needle assembly. Inflammation is evaluated based on appearance changes of the implantation sites including reddening, swelling, and scab formation. After the specimens were injected, the two mice were sacrificed after 34 days (short term) and 97 days (long term), respectively. Tissues include skin, liver, pancreas, and spleen were collected from both mice and fixed in 10% buffered formalin before processed routinely and stained for histological examination.3. Results and discussion
3.1 BSA hydrogel
The structural complexity at quaternary level makes BSA a globular protein suitable for the heat-induced self-healing hydrogel system. It has been known that by modification of the delicate interplay between electrostatic, hydrophobic, hydrogen bonding, and other interactions, the three-dimensional structure of a protein can be changed.26,27 Thermal induced denaturation of BSA can lead to protein conformational changes for gelation24,28,29 (Fig. 1). Firstly, heating induces part of the hydrophobic groups buried in the protein core at ambient temperatures become exposed during the initial protein unfolding.30–32 Then hydrophobic effect leads to non-covalent interactions between unfolded protein molecules to form fine stranded networks.28,30,31 | ||
| Fig. 1 (A) The right vial contains thermal-induced BSA (30 wt%) hydrogel and the left vial contains 30% BSA solution in phosphate buffer; (B) inversed vials contain 30% BSA solution (left) and thermal-induced BSA hydrogel (right). | ||
CD is employed to analyze the conformational changes in BSA during heating process. The CD spectra of BSA in PB after heating from 20–100 °C are shown in Fig. 2. The characteristic of the α-helix structure of BSA typically shows two negative bands centered at 208 nm and 222 nm. With heating temperature lower than 60 °C, it barely changes in the CD spectra, indicating that the secondary structure of BSA is relatively stable when the temperature increases from 20 °C to 60 °C. However, the negative bands decrease considerably with temperature higher than 60 °C. The changes in CD spectra reflect the decrease in the helicity of BSA, implying that high temperature led to the un-folding of the protein molecules. The CD spectra are usually presented in terms of mean residue ellipticity (MRE) with unit deg cm2 dmol−1, which can be calculated as follow:33,34

Where MRE208 is the MRE value at 208 nm; 4000 is the MRE of the β-form and random coil conformation at 208 nm; and 33
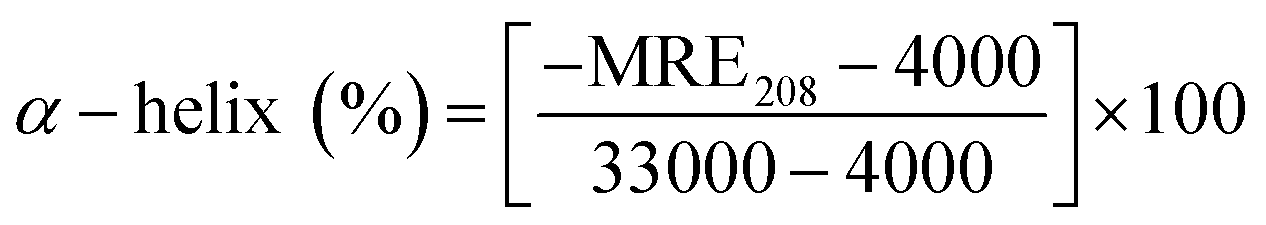 000 is the MRE of a pure α-helix at 208 nm.35
000 is the MRE of a pure α-helix at 208 nm.35
 | ||
| Fig. 2 CD spectra of 0.003 wt% BSA in 20 mM pH 7.4 PB solution after incubated at different temperature (20, 30, 40, 50, 60, 70, 80, 90, and 100 °C, respectively). | ||
As the heating temperature increased from 20 °C to 60 °C, the calculated α-helix content slightly decreased from 41.29% to 40.08%. The negligible change in helicity of the protein indicates that the secondary structure of BSA barely changes during the heating process from 20 °C to 60 °C. This result is in accord with the observation that the gelation temperature is above 60 °C in Fig. 2. However, the α-helix content decreased dramatically from 37.26% to 32.56%, 28.73%, and 22.50% when the heating temperature increased from 70 °C to 80 °C, 90 °C, and 100 °C, respectively. The decreases in α-helix content reveal that heating over 60 °C distorts the secondary structure of the BSA.
ATR-FTIR technique was also employed to study the gelation mechanism of aqueous BSA solution. Fig. 3 shows IR spectra of BSA powder and lyophilized BSA hydrogels with the same dry weight (10 mg). During the thermal-induced denaturation of BSA, mild changes in protein size and shape lead to some of the hydrophobic groups in the protein core exposed to environment, and then hydrophobic effects play an important role for aggregation of BSA to form networks as gel.31 At the secondary structure level, there is a partial loose of the α-helix and an increase in antiparallel β-sheet attributing to the formation of non-native β-sheet associated with aggregation.30,32,36–38 At a molecular level, the number of hydrogen bonds increases after gelation and it increases with the increase of BSA concentration. This claim is clearly supported by the increased intensity in 3000–3600 cm−1 region, which attributes to increasing N–H groups in appearance of carboxyl groups.39 In addition, the peak intensities of both amide I and amide II bands increased due to the increased number of hydrogen bonds.40 To further assess the nature of the interaction within the network of the hydrogel, 30% BSA hydrogels were tested in different environment and the results are shown in Fig. S1.† The thermal-induced BSA hydrogels were stable in acidic (1 M HCl) and physiological (20 mM PBS at pH 7.4) conditions. However, the thermal-induced BSA hydrogels immersed in basic condition (1 M NaOH) or in 8 M urea were fully degraded with time, indicating that non-covalent and probably hydrogen bonding is one of the primary mechanisms in the hydrogel formation too. Additionally, hydrogel samples submerged in 10% SDS turned into transparent and swelled significantly, suggesting that hydrophobic interactions also play a primary role in the gel network formation. Although one cannot rule out the contributions from ionic interaction, electrostatic interaction, etc., these results support that both hydrogen bonds and hydrophobic interaction play important roles in maintaining hydrogel network.
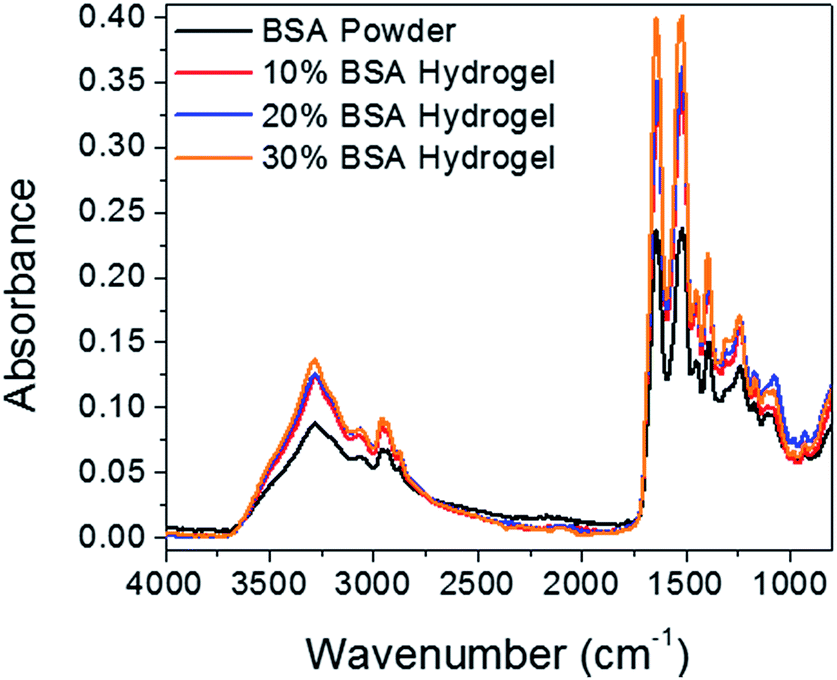 | ||
| Fig. 3 ATR-FTIR spectra of BSA powder and lyophilized thermal-induced BSA hydrogel in concentrations of 10%, 20%, and 30%, respectively. | ||
3.2 Gelation of BSA aqueous solution
BSA dissolved in PB at room temperature shows clear yellow color. With the temperature increased, little change occurred in the aqueous BSA solution until it reached the gelation temperature. After incubating at gelation temperatures for 20 min, transparent hydrogels with slight yellow color were formed. With the temperature increased above the gelation temperature, turbidities were observed visually and the transparent yellow color of the hydrogels gradually turned to opaque white color. The sol–gel transition is irreversible and it maintained the hydrogel structure when the temperature cooled back to room temperature. The gelation temperature was measured by dynamic rheological analysis. Storage modulus (G′) and loss modulus (G′′) of different concentrations of BSA solutions were recorded from 20 °C to 100 °C with an increment of 2 °C and a 20 min equilibration time. As shown in Fig. 4A, at low temperatures G′′ is larger than G′, suggesting the “liquid-like” behavior of the 30% BSA solution. As the temperature increases, G′ gradually increases to a value larger than G′′, implying that the sample becomes more “solid-like”. Although the exact crossover point between G′ and G′′ may depend on the test oscillation frequency, for simplicity, the “gelation temperature” is defined as the temperature at which the G′ is congruent to G′′. As the BSA concentration increased, the gelation temperature of aqueous solution decreased (Fig. 4B). BSA aqueous solution with a concentration lower than 10% showed no sol–gel transition even heated at 100 °C. By adjusting BSA concentration and the heating temperature, the transparency as well as the mechanical properties of the hydrogel can be easily manipulated. | ||
| Fig. 4 (A) G′ and G′′ of a 30% BSA solution as the temperature was increased from 20 °C to 100 °C with an increment of 2 °C and an equilibration time of 20 min at each temperature. Strain amplitude: 1%; frequency: 6.3 rad s−1. (B) Gelation temperature of BSA solutions at different concentrations (10%, 15%, 20%, 25% and 30%) with a negligible standard deviation (triplicate experiments). | ||
3.3 Morphology of BSA hydrogel
The morphology of BSA hydrogel was observed by FESEM. In order to maintain the structures of hydrogels, all freshly prepared specimens were lyophilized for 24 h after frozen in liquid nitrogen. The SEM images of BSA hydrogels prepared with different BSA concentration (Fig. 5) demonstrated that the network of the hydrogel consists of open and interconnected porous structures. The pore sizes and crosslinking densities varied with BSA concentration. In general, a lower concentration of BSA led to a larger pore size in the hydrogels. SEM image of 10% BSA hydrogel sample displays a near layered structure, while images of 20% and 30% BSA hydrogel show a compact and porous structure. Overall, the pore diameter was in the range of 1 to 20 μm. This study indicates that the pore size, porosity and interconnectivity of the hydrogel can be tuned by adjusting the BSA concentration, thus endowing the flexibility in the preparation of BSA hydrogel with controllable properties. | ||
| Fig. 5 FESEM images of lyophilized BSA hydrogels with BSA concentration of 10% (a), 20% (b), and 30% (c), respectively. | ||
3.4 Mechanical analysis – compressive and tensile test
The mechanical properties of a biomaterial are a main concern for many practical applications. Therefore, compressive analysis was carried out to explore the relationship between mechanical strength and BSA concentration of the hydrogel, while tensile tests were conducted to study how temperature influences the mechanical strength of the hydrogel. Fig. 6 depicts the stress–strain curves for BSA hydrogels with different BSA concentration prepared under the same temperature of 80 °C. These specimens exhibit similar stress–strain curves, where approximately 60% strain change occurs as the stress on the sample increases from 0 to about 0.2 MPa and slight strain change as the stress increases above 0.2 MPa. The compression displacement of 20% BSA hydrogel sample (82.05%) is higher than 25% BSA hydrogel (78.43%) and 30% BSA hydrogel (74.06%) samples. This can be ascribed to the structure and porosity difference between hydrogels with different BSA concentration. A comparison of stress–strain curves among BSA hydrogels with different BSA concentration indicates that hydrogels with higher BSA concentrations are stronger than the ones with lower BSA concentration. In addition, the breaking point of all samples cannot be observed up to 18 N. | ||
| Fig. 6 Stress–strain curves of compressive test for 20%, 25%, and 30% BSA hydrogels formed at 80 °C, respectively. | ||
Tensile characterization was further conducted using 30% BSA hydrogels prepared at different incubating temperature (70 °C, 75 °C and 80 °C) and the results demonstrates that a higher incubation temperature leads to increased maximum stress required to break the hydrogel but less elongation capability (Fig. 7). The fracture stress in 30% BSA hydrogels increases about 16-fold (from 3.97 kPa to 63.77 kPa) with the incubation temperature increasing from 70 °C to 80 °C, while the flexibility of the hydrogel dropped dramatically. The specimen incubated at 70 °C can be elongated about 2.5-fold before fracture while the specimen incubated at 80 °C can only stand less than 50% elongation. These results suggest that the flexibility as well as the strength of the hydrogel can be controlled by changing the BSA concentration or the incubation temperature of the hydrogel.
 | ||
| Fig. 7 Stress–strain curves of tensile test for 30% hydrogels formed at 70 °C, 75 °C and 80 °C, respectively. | ||
3.5 Self-healing capability
Different from other denaturation methods by which a protein completely unfolds, the size and shape of protein only mildly perturbed in the as-developed thermal-induced denaturation.31 As a result, the multiple molecular interactions contributing to thermal-induced gel formation can be employed for self-healing purpose at the cut surfaces of the as-prepared BSA hydrogel. As a demonstration, a rod-shaped BSA was cut by a razor into multiple segments. By simply keeping the cut surfaces in contact at 80 °C for 60 min the mechanical damage in the hydrogel could be self-healed (Fig. 8A), and this self-healing process is repeatable (Fig. 8B). | ||
| Fig. 8 (A) First-time self-healing of BSA hydrogel: BSA hydrogel (original) was cut to four pieces using a razor and then self-healed by keeping the cut surfaces in contact for 60 min at 80 °C; (B) repetitive self-healing of BSA hydrogel: self-healed BSA hydrogel in (A) was cut again to four pieces then self-healed by keeping the cut surfaces in contact for 60 min at 80 °C, demonstrating repetitive self-healing property. | ||
Similarly, the self-healing behavior after mechanical damage recovery was observed in BSA hydrogels consisting of different fluorophores. Fluorescein and eosin Y disodium salt were incorporated in the precursor solution to prepare the hydrogels, respectively. The hydrogel with fluorescein shows yellow color under white light and green fluorescence under UV light, while the hydrogel with eosin Y disodium salt shows red color under white light and orange color under UV light (Fig. 9A and B). The eosin Y concentration is too high to self-quench its fluorescence, thus resulting in weak fluorescence. Both hydrogels labelled with different fluorophores were cut by a razor into three pieces (Fig. 9C and D) and blend for the healing process described before when connecting the three pieces of protein hydrogel into one. It shows that BSA hydrogels consisting of different fluorophore can be combined by joining two different cut surfaces (Fig. 9E–H). Strong attachment between hydrogel pieces labelled with different fluorophore was also observed (Fig. 9E–H) with clear demarcation. The self-integration process of simply adjoining the cross-sectional surfaces of different BSA hydrogels provides potential applications in design and fabrication of novel multi-segment biomaterials with stimuli-sensitivity and programmed controlled-release property.
 | ||
| Fig. 9 BSA hydrogels incorporated with fluorescein and eosin Y disodium salt under white light (A) and UV light (B); BSA hydrogels with different fluorophores cut into three pieces by a razor under white light (C) and UV light (D); self-healed blended BSA hydrogels under white light (E and G) and UV light (F and H). | ||
The compressive tests were performed to study the mechanical properties of the original and repaired hydrogels. Fig. 10 depicts the load–strain curves for original and repaired samples after different healing times (20, 40 or 60 min) at 80 °C. All the samples exhibit similar load-strain curves, a low rate up to approximately 60% strain was observed when the loads on the sample increased from 0.01 to about 3 N and then more rapidly rate between 60% and 80% strain in the range from about 3 to 18 N. There is no sharp peak observed in all curves, indicating that there is no huge disfigurement appeared in both original and repaired hydrogels during compression. The maximum strain slightly decreased from 86.81% to 86.71%, 84.75% and 82.87% after the hydrogels were self-healed for 20, 40, and 60 min, respectively. Increased hydrophobic interaction, hydrogen bonding and a higher degree of denaturation of BSA induced by thermal treatment during self-healing process may increase the stiffness of the self-healed hydrogel, thus leading to the observed slightly decrease of the maximum strain.
 | ||
| Fig. 10 Load–strain curves for the as-prepared and the self-healed hydrogels. | ||
Tensile tests were performed to quantitatively evaluate the self-healing properties of the original and repaired BSA hydrogels after different healing times (20, 40, and 60 min). The healing process was performed as described before and the rod-shaped samples were cut into films (6 mm length × 4 mm width × 1 mm thickness) which also contained the healed surface in the middle. Fig. 11 shows that stress–strain curves of the as-prepared and self-healed hydrogels (healed with different time) behave differently. Samples healed after 20 and 40 min at 80 °C can only recover 17% and 36% of break strength of the original hydrogel, respectively. However, the tensile strength and break strain of the sample healed for 60 min at 80 °C can reach over 100% of the original strength and elongation abilities and then subsequently broke along the same plane as the original cut. The self-healing process could be attributed to the reformation of the hydrophobic interaction and hydrogen bonding between the available carbonyl oxygen and NH2 groups on the cut surfaces.
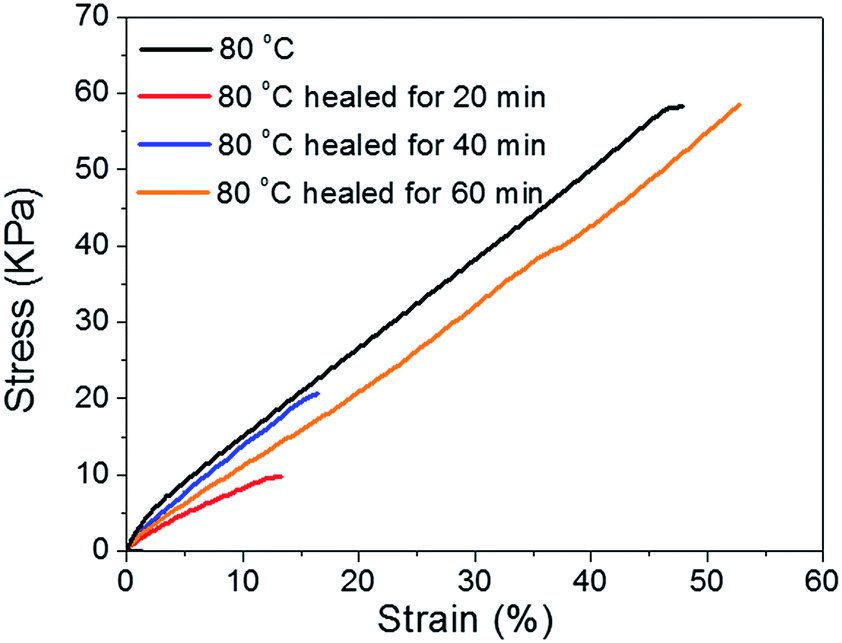 | ||
| Fig. 11 Stress–strain curves for as-prepared and self-healed hydrogels. | ||
3.6 Cytotoxicity analysis and histomorphometry near the injection site
In vitro cytotoxicity tests were first performed to forecast in vivo inflammatory responses.6 The colorimetric assay was used to evaluate the cell viability based on increase of absorption at 450 nm after adding CCK-8.41 Cytotoxicity tests were performed before in vivo studies in order to explore if there is any potentially dangerous chemicals in the hydrogel.42 Thus, cytotoxicity in vitro is a necessary step before tests in animal models to minimize the acute phase in animals during in vivo studies.43 The result of the in vitro cell viability assay (Fig. 12) indicated that the survival rate varied with the concentration of the hydrogel extract solutions. The average of cell survival percentage and the standard deviation for each concentration were generated from a sample size of n = 3. More than 90% of the cells remained alive in all tests. Even when the cells were exposed to the hydrogel extract solution with the concentration as high as 1%, nearly 90% cells were still alive, indicating the low toxicity of the thermal-induced BSA hydrogel. | ||
| Fig. 12 The survival rate of BSA hydrogel extract solution in different concentration. | ||
The immunogenicity of a biomaterial is crucial for its success in in vivo biomedical applications. After the subcutaneous administration of the BSA hydrogel into immunocompetent mice, both short-term (34 days) and long-term (97 days) investigations were performed to identify the histocompatibility of the hydrogel. The inflammatory reactions were evaluated by color change (reddening), swelling, and scab formation of subcutaneous injection site.44 Acute inflammation is the immediate response after injection of a foreign material, involving dilation of vessels and excess of blood.45,46 Therefore, reddening was taken as the most important index to assess the acute inflammatory reaction after injection. The results shown in Fig. S2† suggest that the reddening due to bleeding during injection disappeared after 1 day, indicating the negligible acute inflammatory response for this material.
Histological examination results showed that a mild hyperkeratosis on the epidermis with a moderate number of hair follicles and adnexa in the dermis without other lesions was observed in skin tissues (Fig. 13A and E). Fig. 14 shows that there is a little hydrogel residue after 34 days of injection (Fig. 14A), while no hydrogel left after 97 days (Fig. 14B), demonstrating that this protein based hydrogel could be gradually digested, offering an excellent biodegradable matrix. Additionally, there were no significant lesions in liver (Fig. 13D and H) and pancreas tissues (Fig. 13C and G). There were numerous germinal centers forming the white pulp with a marked lymphoid hyperplasia in the center in spleen tissues (Fig. 13B and F). As the mice were immunocompetent, the red pulp containing extramedullary hematopoesis with a mild hemosiderosis could be considered as normal.
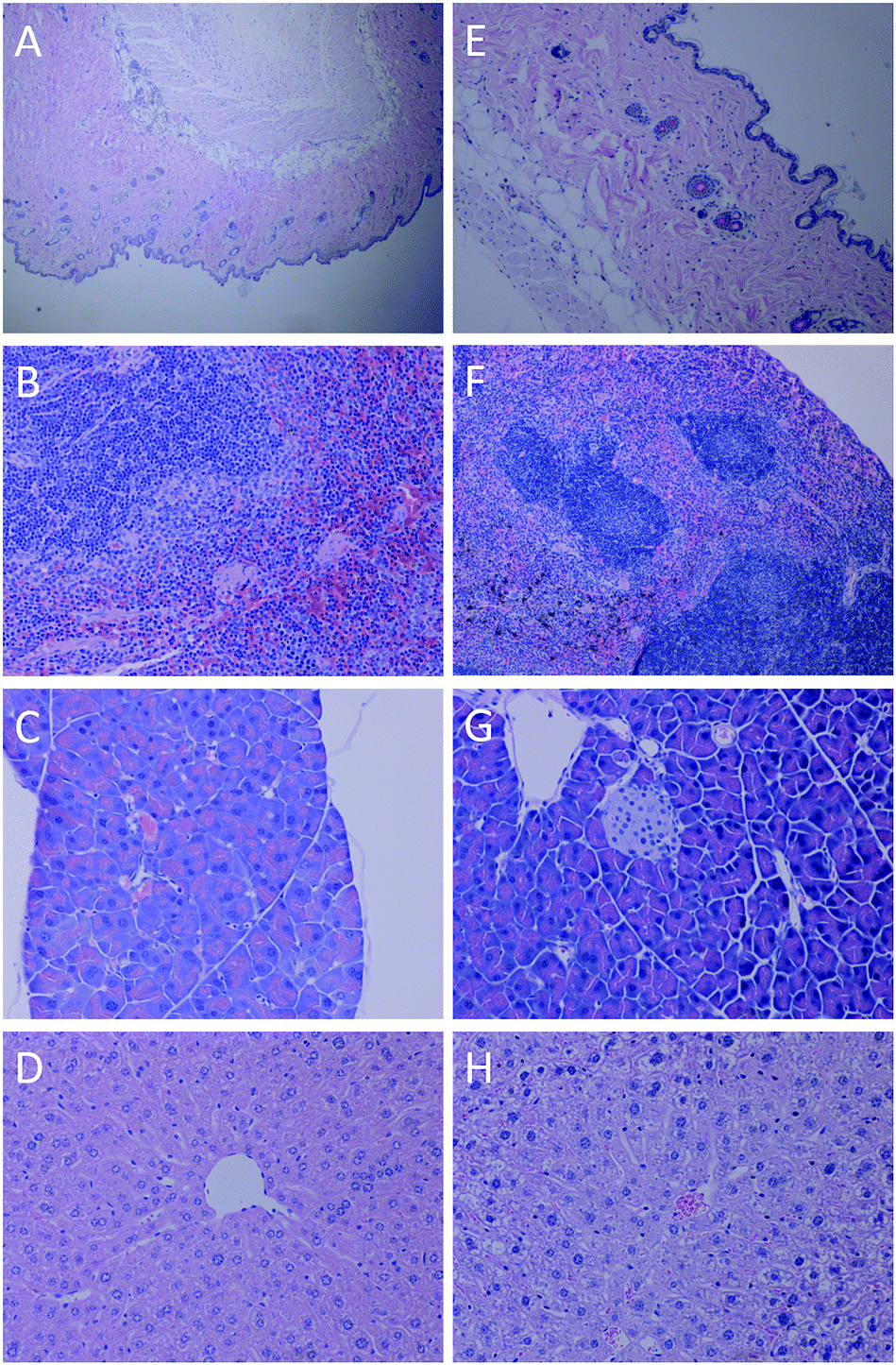 | ||
| Fig. 13 Images of stained tissues of mouse sacrificed after 34 days: skin tissue (40×) (A); spleen tissue (100×) (B); pancreas tissue (100×) (C); liver tissue (100×) (D), respectively; and images of stained tissues of mouse sacrificed after 97 days: skin tissue (100×) (E); spleen tissue (100×) (F); pancreas tissue (100×) (G); liver tissue (100×) (H), respectively. | ||
 | ||
| Fig. 14 Skin tissue (100×) focusing on subcutaneous area in the injection site after 34 days (A) and 97 days (B), respectively. | ||
4. Conclusion
Thermal-induced BSA hydrogel was systematically investigated in this study. By controlling the heating temperature and the concentration of BSA, the mechanical strength of the as-prepared hydrogel could be manipulated. With the help of heating, the damaged protein hydrogel shows repeatable self-healing property, which was reported for the first time. Tensile tests were used to quantitatively evaluate the self-healing efficiency, showing nearly 100% recovery. Both in vitro and in vivo tests were systematically carried out to demonstrate the good biocompatibility of the hydrogel. The in vivo study also shows that the thermal-induced BSA hydrogel could be fully degraded with negligible acute inflammatory response and possesses excellent long-term biocompatibility. The biodegradable hydrogel contain self-healing property would bring light into the design of new class of controlled-release drug delivery system in which drug release could be manipulated both spatially and temporally.Acknowledgements
We acknowledge financial support from National Science Foundation (NSF). We also appreciate Connecticut Veterinary Medical Diagnostic Laboratory in the help of histological analysis.Notes and references
- M. K. Nguyen and D. S. Lee, Macromol. Biosci., 2010, 10, 563–579 CrossRef CAS PubMed.
- L. Oss-Ronen and D. Seliktar, Adv. Eng. Mater., 2010, 12, B45–B52 CrossRef.
- J.-C. Gayet and G. Fortier, J. Controlled Release, 1996, 38, 177–184 CrossRef CAS.
- Y. Zhang, B. Yang, X. Zhang, L. Xu, L. Tao, S. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- S. Kızılel, E. Sawardecker, F. Teymour and V. H. Pérez-Luna, Biomaterials, 2006, 27, 1209–1215 CrossRef PubMed.
- X. Ma, J. Deng, Y. Du, X. Li, D. Fan, C. Zhu, J. Hui, P. Ma and W. Xue, J. Mater. Chem. B, 2014, 2, 2749–2763 RSC.
- F. Brandl, N. Hammer, T. Blunk, J. Tessmar and A. Goepferich, Biomacromolecules, 2010, 11, 496–504 CrossRef CAS PubMed.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS PubMed.
- N. Huebsch, C. J. Kearney, X. Zhao, J. Kim, C. A. Cezar, Z. Suo and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9762–9767 CrossRef CAS PubMed.
- C.-C. Lin and K. S. Anseth, Pharm. Res., 2009, 26, 631–643 CrossRef CAS PubMed.
- R. Censi, T. Vermonden, H. Deschout, K. Braeckmans, P. di Martino, S. C. De Smedt, C. F. van Nostrum and W. E. Hennink, Biomacromolecules, 2010, 11, 2143–2151 CrossRef CAS PubMed.
- G. J. Im, S. Y. Chae, K. C. Lee and D. S. Lee, J. Controlled Release, 2009, 137, 20–24 CrossRef PubMed.
- J. K. Tessmar and A. M. Göpferich, Adv. Drug Delivery Rev., 2007, 59, 274–291 CrossRef CAS PubMed.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed.
- H. Park, J. S. Temenoff, Y. Tabata, A. I. Caplan and A. G. Mikos, Biomaterials, 2007, 28, 3217–3227 CrossRef CAS PubMed.
- Z. Xie, Y. Zhang, L. Liu, H. Weng, R. P. Mason, L. Tang, K. T. Nguyen, J. T. Hsieh and J. Yang, Adv. Mater., 2014, 26, 4491–4496 CrossRef CAS PubMed.
- J. S. Temenoff and A. G. Mikos, Biomaterials, 2000, 21, 2405–2412 CrossRef CAS PubMed.
- H. Tan and K. G. Marra, Materials, 2010, 3, 1746–1767 CrossRef CAS.
- S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Mol. Biol., 1998, 5, 827–835 CAS.
- X. Ma, D. Hargrove, Q. Dong, D. Song, J. Chen, S. Wang, X. Lu, Y. K. Cho, T.-H. Fan and Y. Lei, RSC Adv., 2016, 6, 50091–50099 RSC.
- X. Ma, X. Sun, D. Hargrove, J. Chen, D. Song, Q. Dong, X. Lu, T.-H. Fan, Y. Fu and Y. Lei, Sci. Rep., 2016, 6, 19370 CrossRef CAS PubMed.
- X. Ma, T. Wang, D. Song, D. Hargrove, Q. Dong, Z. Luo, J. Chen, X. Lu, Y. Luo and T.-H. Fan, ACS Biomater. Sci. Eng., 2016 DOI:10.1021/acsbiomaterials.6b00048.
- E. Gelamo and M. Tabak, Spectrochim. Acta, Part A, 2000, 56, 2255–2271 CrossRef.
- K. Baler, R. Michael, I. Szleifer and G. A. Ameer, Biomacromolecules, 2014, 15, 3625–3633 CrossRef CAS PubMed.
- P. L. Nasatto, F. Pignon, J. L. Silveira, M. E. R. Duarte, M. D. Noseda and M. Rinaudo, Polymers, 2015, 7, 777–803 CrossRef CAS.
- K. Baler, O. Martin, M. Carignano, G. Ameer, J. Vila and I. Szleifer, J. Phys. Chem. B, 2014, 118, 921–930 CrossRef CAS PubMed.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153–203 CrossRef CAS PubMed.
- C. Giancola, C. De Sena, D. Fessas, G. Graziano and G. Barone, Int. J. Biol. Macromol., 1997, 20, 193–204 CrossRef CAS PubMed.
- M. Murata, F. Tani, T. Higasa, N. Kitabatake and E. Doi, Biosci., Biotechnol., Biochem., 1993, 57, 43–46 CrossRef CAS.
- A. Clark, G. Kavanagh and S. Ross-Murphy, Food Hydrocolloids, 2001, 15, 383–400 CrossRef CAS.
- W. S. Gosal and S. B. Ross-Murphy, Curr. Opin. Colloid Interface Sci., 2000, 5, 188–194 CrossRef CAS.
- V. Militello, C. Casarino, A. Emanuele, A. Giostra, F. Pullara and M. Leone, Biophys. Chem., 2004, 107, 175–187 CrossRef CAS PubMed.
- X. L. Lu, H. C. Yang, H. Wu and A. X. Hou, J. Mol. Biomarkers Diagn., 2012, 2, 126 Search PubMed.
- H. Xu, N. Yao, H. Xu, T. Wang, G. Li and Z. Li, Int. J. Mol. Sci., 2013, 14, 14185–14203 CrossRef PubMed.
- H. Gao, L. Lei, J. Liu, Q. Kong, X. Chen and Z. Hu, J. Photochem. Photobiol., A, 2004, 167, 213–221 CrossRef CAS.
- J. I. Boye, I. Alli and A. A. Ismail, J. Agric. Food Chem., 1996, 44, 996–1004 CrossRef CAS.
- G. Navarra, D. Giacomazza, M. Leone, F. Librizzi, V. Militello and P. L. San Biagio, Eur. Biophys. J., 2009, 38, 437–446 CrossRef CAS PubMed.
- C. Honda, H. Kamizono, T. Samejima and K. Endo, Chem. Pharm. Bull., 2000, 48, 464–466 CrossRef CAS PubMed.
- J. Grdadolnik and Y. Maréchal, Biopolymers, 2001, 62, 40–53 CrossRef CAS PubMed.
- H.-Y. Park, I.-H. Song, J.-H. Kim and W.-S. Kim, Int. J. Pharm., 1998, 175, 231–236 CrossRef CAS.
- A. Kroll, M. H. Pillukat, D. Hahn and J. Schnekenburger, Eur. J. Pharm. Biopharm., 2009, 72, 370–377 CrossRef CAS PubMed.
- M. Garle, J. Fentem and J. Fry, Toxicol. In Vitro, 1994, 8, 1303–1312 CrossRef CAS PubMed.
- G. J. Harry, M. Billingsley, A. Bruinink, I. L. Campbell, W. Classen, D. C. Dorman, C. Galli, D. Ray, R. A. Smith and H. A. Tilson, Environ. Health Perspect., 1998, 106, 131 CrossRef CAS PubMed.
- Y. J. Heo, H. Shibata, T. Okitsu, T. Kawanishi and S. Takeuchi, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13399–13403 CrossRef CAS PubMed.
- B. Říhová, Adv. Drug Delivery Rev., 1996, 21, 157–176 CrossRef.
- B. Říhová, Adv. Drug Delivery Rev., 2000, 42, 65–80 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra11239k |
| This journal is © The Royal Society of Chemistry 2016 |