
Catalyst-free and selective synthesis of 2-aminothiophenes and 2-amino-4,5-dihydrothiophenes from 4-thiazolidinones in water†
Fanxun Zenga,
Pengjian Liua,
Xusheng Shaoa,
Zhong Liab and
Xiaoyong Xu*ab
aShanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, China. E-mail: xyxu@ecust.edu.cn
bShanghai Collaborative Innovation Center for Biomanufacturing Technology, 130 Meilong Road, Shanghai 200237, China
First published on 16th June 2016
Abstract
2-Aminothiophenes and 2-amino-4,5-dihydrothiophenes were selectively and conveniently synthesized from 4-thiazolidinone derivatives. The meaningful product 10m was efficiently synthesized, which is a commonly-used intermediate for preparing olanzapine. The present method holds many advantages, such as easy operation, high yields, is catalyst-free and uses water as the solvent.
Introduction
Thiophenes are widely used in drug design1 and can be found in a number of marketed drugs such as olanzapine 1, plavix 2 and cymbalta 3 (Fig. 1).2 Continuous studies on thiophenes are also involved in dye chemistry,3 materials science,4 electronic and optoelectronic devices,5 biodiagnostics,6 block copolymer self-assembled superstructures, and conductivity-based sensory devices.7 As derivatives of thiophenes, dihydrothiophenes are also privileged scaffolds which present in natural products, bioactive molecules and synthetic intermediates.8 For example, 4,5-dihydrothiophene-3-carbonitrile 4 (Fig. 1)9 exhibit antibacterial and antifungal properties and 2-amino-4,5-dihydrothiophenes (ADHTs) are used as starting materials for the synthesis of partially hydrogenated thieno[2,3-b]pyridines10 and pyrimidines.11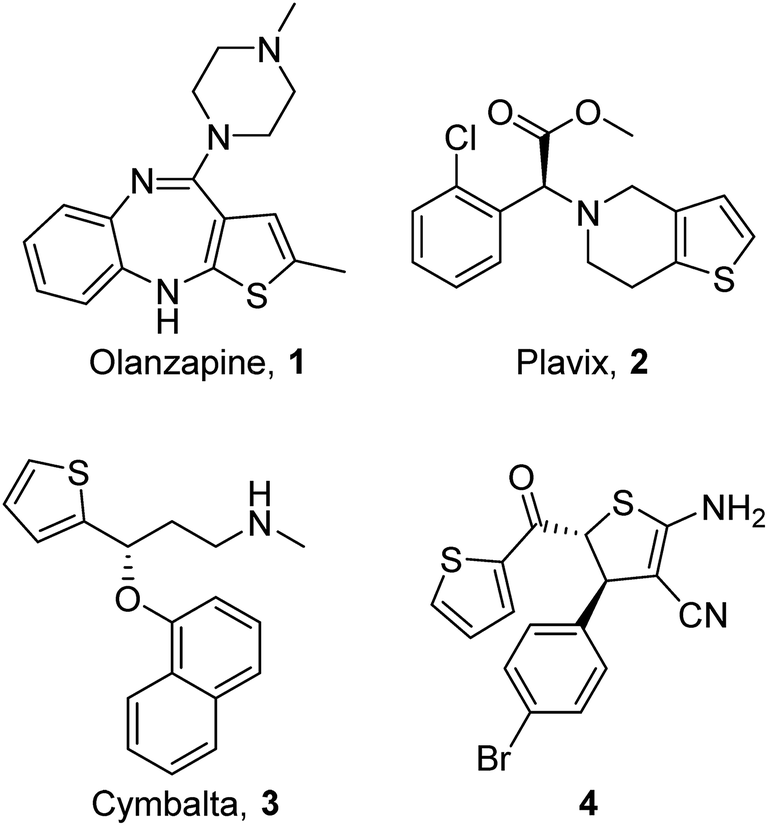 | ||
| Fig. 1 Examples of bioactive thiophenes and dihydrothiophenes. | ||
A variety of synthetic methods have been developed for the preparation of 2-aminothiophenes (ATs), of which the most attractive route is Dieckmann or Thorpe–Ziegler cyclization of thioacetals in basic medium (Scheme 1).12 In addition, Brandsma explored a one-pot methodology using acetylenes and isothiocyanates for constructions of 5-substituted 2-aminothiophenes.13 Wang and co-workers disclosed the efficient synthesis of 3-substituted-2-aminothiophenes from substituted allyl benzotriazoles and isothiocyanates.14 Then, ATs obtained by treatment of arylaminothieno-oxobutanamides with Lawesson's reagent were reported.15 Recently, isothiocyanates were reported to react with electron-deficient allenes16 or Morita–Baylis–Hillman adducts17 to prepare ATs. In general, these synthetic methods have inherent drawbacks such as narrow substrate scope and the use of strong base n-butyllithium. Therefore, the development of concise and efficient methods for the access of ATs is still in high demand.
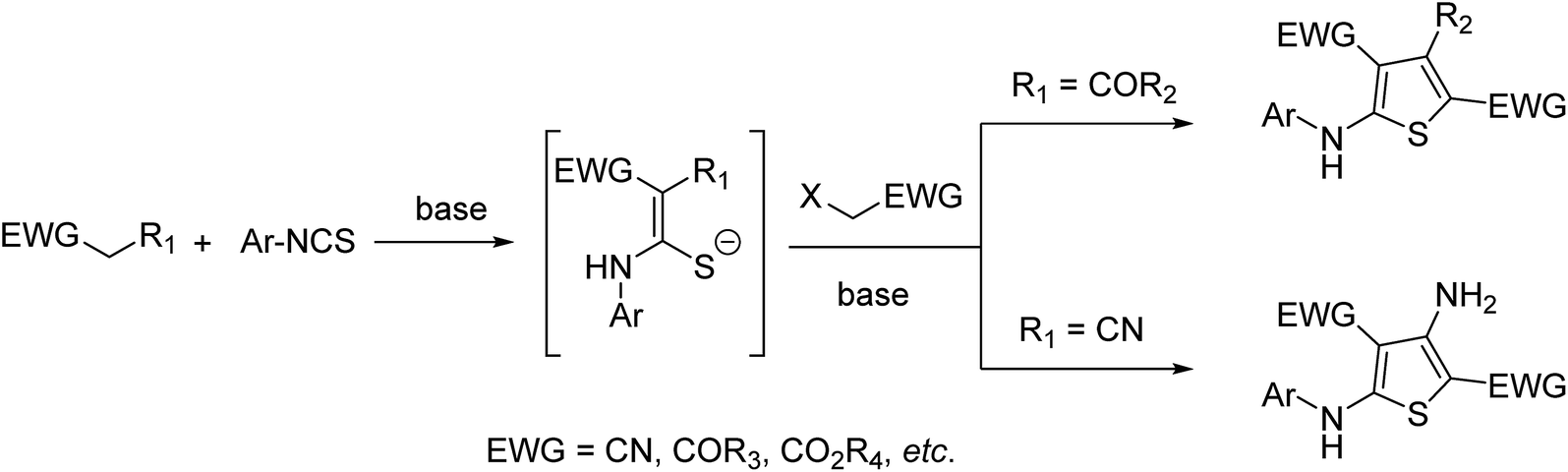 | ||
| Scheme 1 Synthesis of ATs via Dieckmann or Thorpe–Ziegler cyclization of thioacetals. | ||
Although ADHTs have great synthetic value, there are fewer publications on their synthesis in comparison with the detailed study on synthesis of thiophenes. Dotsenko and co-workers reported the synthesis of ADHTs by base-catalyzed condensation of phenacyl thiocyanate with substituted thioamides.18 Shestopalov et al. disclosed a three-component condensation reaction involving aldehydes, cyanothioacetamide and pyridinium or sulfonium ylides for synthesis of ADHTs (Scheme 2, eqn (1) and (2)).19 ADHTs could also be synthesized by a domino reaction involving 1,3-thiazolidinedione, malononitrile and aromatic aldehydes (Scheme 2, eqn (3)).8g More recently, ADHTs were synthesized by Chandrasekaran from doubly activated cyclopropanes using tetrathiomolybdate as the sulfur transfer reagent (Scheme 2, eqn (4)).20 However, these protocols suffer from one or more drawbacks, such as low yields and the narrow scope of substrates.
 | ||
| Scheme 2 Previous strategies for the preparation of ADHTs. | ||
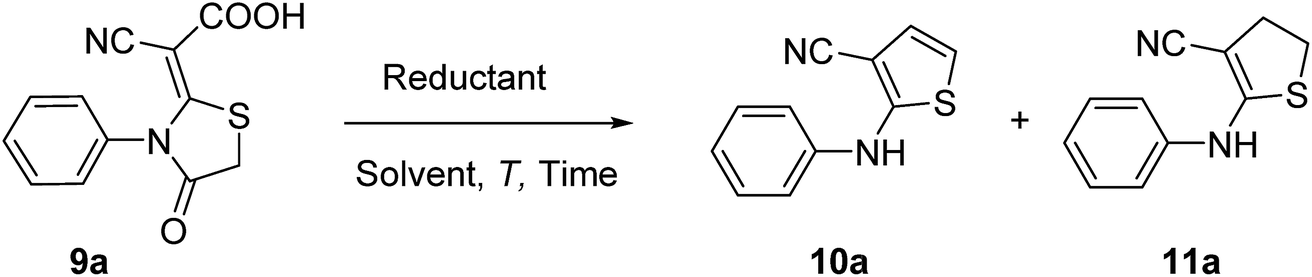
Here, the synthesis of ATs and ADHTs from 4-thiazolidinone derivatives was reported. At the beginning, we intended to selectively reduce the carboxyl group of 4-thiazolidinone derivatives 9 using NaBH4,21 because 4-thiazolidinones possess broad biological activities.22 Unfortunately, no target product was obtained after many attempts. To our delight, interesting products ATs and ADHTs were detected. Then, the optimized reaction conditions and the scope of substrates were explored and the possible mechanism was also proposed.
Results and discussions
Starting materials aryl isothiocyanates 5 (ref. 23) and key intermediates 4-thiazolidinone derivatives 8 (ref. 21e) were easily obtained according to reported procedures. The corresponding carboxylic acids derivatives 9 were prepared by cleavage of tert-butyl with TFA in DCM (Scheme 3). | ||
| Scheme 3 Preparation of 4-thiazolidinone derivatives 9 from aryl isothiocyanates 5. | ||
Initially, the reaction conditions were optimized (Table 1) for the direct synthesis of AT using N-phenyl 4-thiazolidinone (9a) as a model substrate. Various reductants and solvents were screened at room temperature, and the results are presented in Table 1. Among the various reductants tested, NaBH4 was found to be superior in terms of yield of 10a (Table 1, Entry 1–4). Among the solvents screened, water was found to give the best result (Table 1, Entry 6). Other organic solvents, such as THF, MeOH and EtOH, afforded the product in relatively lower yields (Table 1, Entries 1, 2 and 5). The influence of the amount of reductants was then investigated, and 2.0 equiv. of NaBH4 was the most effective. Moreover, it was found that the yield of AT decreased and ADHT would occur when 3.0 equiv. of NaBH4 was used (Table 1, Entry 10). The effect of temperature was also studied. The yield of 10a increased to a remarkable 87% when the temperature was decreased to 15 °C (Table 1, Entry 11). Further decreasing the temperature to 5 °C led to significant reduction of the yield and extended reaction time (48 h) (Table 1, Entry 12). Therefore, Entry 11 was selected for the optimal reaction conditions. In order to investigate the relationship between the amount of NaBH4 and the products ratio, the amount of NaBH4 was further increased. Gratifyingly, the yield of 11a increased to 54% when 10.0 equiv. of NaBH4 was employed (Table 1, Entry 15). We next examined the effect of base in water on the reaction. Saturated aqueous NaHCO3, saturated aqueous KHCO3, 1 M Na2CO3, 1 M K2CO3, 1 M NaOH and 1 M KOH were tried, but the yield of 11a was not improved with AT as the main product (Table 1, Entry 15 vs. Entries 17–22). At 10.0 equiv. of NaBH4, MeOH was investigated again and the result was similar with that of Entry 2. Obvious improved yield and ratio were observed upon using EtOH as solvent (Table 1, Entries 15 vs. Entry 24). The similar yield was obtained when KBH4 was used in water (Table 1, Entry 27), but the ratio is unsatisfied. To further increase the yield of 11a, the reactions were carried out at different temperatures (Table 1, Entry 26–31). The yield of 11a increased to 85% when the temperature was increased to 60 °C and 2 mL H2O was used (Table 1, Entry 30). Further increasing the temperature to 80 °C did not give better results (Table 1, Entry 31). The yield of 11a decrease to 70% when 5.0 equiv. of KBH4 was employed (Table 1, Entry 32). Based on the above observation, Entry 30 was selected for the optimal reaction conditions.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reductant (equiv.) | Solvent | Temp (°C) | Time (h) | Yieldc (%) 10a | Yieldc (%) 11a |
| a Reaction conditions: substrate 9a (0.2 mmol), water (1 mL).b Reaction conditions: substrate 9a (0.2 mmol), water (2 mL).c Yield was determined by UPLC (ultra performance liquid chromatography).d Isolated yield. | ||||||
| 1 | NaBH4 (2.0) | THF | 25 | 24 | 6 | Trace |
| 2 | NaBH4 (2.0) | MeOH | 25 | 24 | 20 | Trace |
| 3 | NaBH3CN (2.0) | MeOH | 25 | 24 | Trace | Trace |
| 4 | LiAlH4 (2.0) | THF | 25 | 0.5 | 8 | Trace |
| 5 | NaBH4 (2.0) | EtOH | 25 | 8 | 68 | Trace |
| 6 | NaBH4 (2.0) | H2O | 25 | 8 | 78 | Trace |
| 7 | NaBH4 (1.0) | H2O | 25 | 24 | 8 | Trace |
| 8 | NaBH4 (1.2) | H2O | 25 | 24 | 68 | Trace |
| 9 | NaBH4 (1.5) | H2O | 25 | 18 | 70 | Trace |
| 10 | NaBH4 (3.0) | H2O | 25 | 6 | 62 | 21 |
| 11 | NaBH4 (2.0) | H2O | 15 | 20 | 87 (83d) | Trace |
| 12 | NaBH4 (2.0) | H2O | 5 | 48 | 72 | Trace |
| 13 | NaBH4 (5.0) | H2O | 25 | 6 | 37 | 47 |
| 14 | NaBH4 (7.5) | H2O | 25 | 6 | 29 | 51 |
| 15 | NaBH4 (10) | H2O | 25 | 2 | 25 | 54 |
| 16 | NaBH4 (10) | H2O | 15 | 3 | 33 | 52 |
| 17 | NaBH4 (10) | Satd. aqueous NaHCO3 | 25 | 3 | 69 | 5 |
| 18 | NaBH4 (10) | Satd. aqueous KHCO3 | 25 | 3 | 56 | Trace |
| 19 | NaBH4 (10) | 1 M Na2CO3 | 25 | 3 | 47 | 26 |
| 20 | NaBH4 (10) | 1 M K2CO3 | 25 | 3 | 48 | 15 |
| 21 | NaBH4 (10) | 1 M NaOH | 25 | 0.5 | Trace | Trace |
| 22 | NaBH4 (10) | 1 M KOH | 25 | 0.5 | Trace | Trace |
| 23 | NaBH4 (10) | MeOH | 25 | 4 | 24 | Trace |
| 24 | NaBH4 (10) | EtOH | 25 | 4 | 5 | 62 |
| 25 | KBH4 (10) | EtOH | 25 | 4 | Trace | Trace |
| 26 | KBH4 (10) | H2O | 15 | 3 | 33 | 58 |
| 27 | KBH4 (10) | H2O | 25 | 2 | 27 | 63 |
| 28 | KBH4 (10) | H2O | 40 | 0.5 | 18 | 74 |
| 29b | KBH4 (10) | H2O | 40 | 0.5 | 16 | 77 |
| 30b | KBH4 (10) | H2O | 60 | 0.2 | 6 | 85 (81d) |
| 31b | KBH4 (10) | H2O | 80 | 0.2 | 14 | 77 |
| 32b | KBH4 (5.0) | H2O | 60 | 0.2 | 22 | 70 |
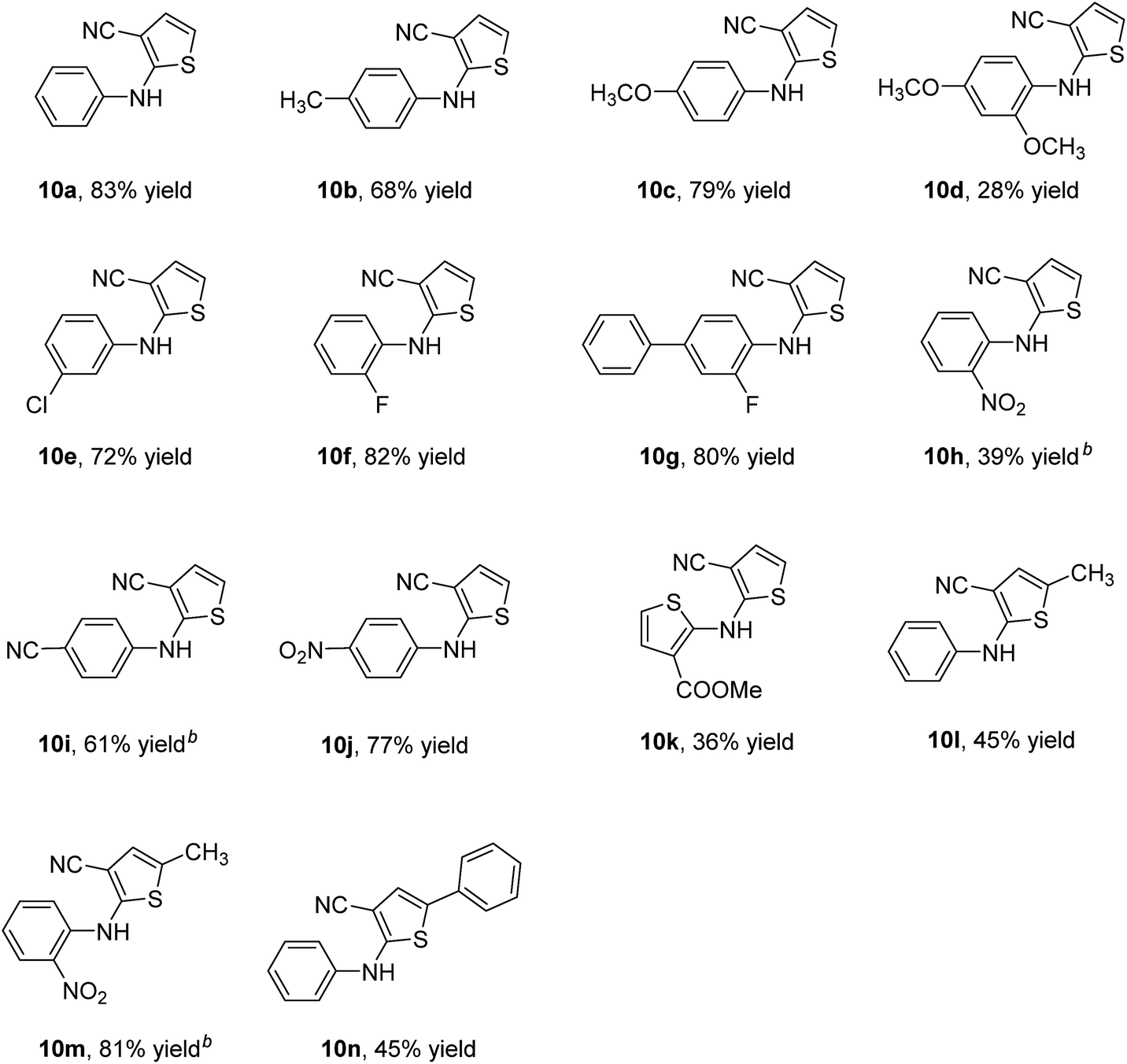
With the optimal conditions established, we explored the substrates scope for synthesis of ATs. The results were summarized in Table 2 and it demonstrated broad substrates scope. The reactions of substrates bearing electron-donating groups on the aromatic ring of 4-thiazolidinones, such as methyl (9b) and methoxy (9c) groups, afforded the corresponding products in good yields. But for the substrate containing an 2,4-dimethoxy group, the yield of 10d decreased rapidly. Meanwhile, the reactions of substrates bearing electron-withdrawing groups on the aromatic ring of 4-thiazolidinones, such as fluoro (9f), chloro (9e), cyano (9i) and nitro (9j) groups, also afforded the corresponding products in good yields. In the case of substrate containing an ortho-nitro (9h) group, only 39% yield was obtained. Heteroaromatic substrate (9k) provided a lower yield. Upon the introducing methyl or phenyl into thiophene ring, the reactions also occurred smoothly to generate corresponding products 10l–10n in moderate to good yields. Most important, the meaningful product 10m could be provided conveniently, which is the commonly-used intermediate of typical antipsychotics olanzapine.24

Then, the scope of substrates to give ADHTs was explored (Table 3). The reactions of substrates bearing electron-donating or electron-withdrawing groups proceeded smoothly to give the expected products 11a–11j in moderate to good yields. In case of the substrates containing 2,4-dimethoxy and 4-nitro groups, the yields of products 11d and 11i decreased slightly, which were different to the reactions of ATs. When benzene ring was replaced with biphenyl, the yield of 11g was only 52%. The decreased water solubility of substrate 9g might cause the lower yield. Heteroaromatic substrate (9k) also performed smoothly to afford product 11k in moderate yield. The yield still remained high when the R2 was the methyl group. But upon changing R2 to phenyl, no desired product was obtained and the major product was the AT 10n in yield of 56%.


Besides 1H, 13C and 19F NMR spectroscopy, the structures of 10g and 11a were confirmed by X-ray diffraction analysis (Fig. 2). The results confirmed that the ADHTs were obtained instead of 2-amino-2,3-dihydrothiophenes. In addition, ADHTs 11 exists amino–imino automerism due to prototropic tautomerism (Scheme 4). This tautomerism can be seen in both 1H, 13C and 19F NMR spectra. X-ray single crystal analysis of 11a revealed that it only exist amino form (Fig. 2). The similar tautomerism was previously studied in the 2-amino-1,3-thiazolidin-4-one derivatives.25
 | ||
| Fig. 2 Single-crystal X-ray diffraction structures of 10g and 11a. | ||
 | ||
| Scheme 4 Tautomers of 11. | ||
A possible reaction mechanism was proposed in Scheme 5. Initially, reduction and deprotonation of 4-thiazolidinone derivatives 9 led to formation of carboxylates A. The followed C–N bond cleavage formed aldehydes B, which underwent intramolecular cyclization to form tetrahydrothiophenes C. Subsequently, tetrahydrothiophenes C were consumed in two paths: a and b. Tetrahydrothiophenes C released CO2 to give hydroxyl compounds D in path a. Finally, hydroxyl compounds D underwent a dehydration process to give ATs 10. In path b, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond was reduced first to form carboxylates E, the second step was decarboxylation of carboxylates E with the followed release of hydroxyl ion, and 2,5-dihydrothiophenes G were given. 2,5-Dihydrothiophenes G can be easily converted to more stable 4,5-dihydrothiophenes 11.
N bond was reduced first to form carboxylates E, the second step was decarboxylation of carboxylates E with the followed release of hydroxyl ion, and 2,5-dihydrothiophenes G were given. 2,5-Dihydrothiophenes G can be easily converted to more stable 4,5-dihydrothiophenes 11.
 | ||
| Scheme 5 Proposed mechanism for the formation of 10 and 11. | ||
Conclusions
A new and efficient method for the synthesis of ATs and ADHTs derivatives was developed using inexpensive and readily available starting materials and reagents. Many advantages, such as easy operation, high yields, using of water as a solvent, catalyst-free and broad substrates scope, make this method absorb more attention.Experimental section
General procedure for the synthesis of tert-butyl-2-cyano-2-(4-oxo-3-arylthiazolidin-2-ylidene)acetates (8a–8n)
Tert-Butyl cyanoacetate (10 mmol) followed by a solution of aryl isothiocyanate 5 (10 mmol) in anhydrous DMF (10 mL) were added to a cold suspension of powdered KOH (20 mmol) in dry DMF (10 mL). The mixture was stirred at room temperature for 0.5 h, then cooled again to 0 °C, treated with a solution of appropriate 2-halogen acyl chloride 7 (15 mmol) in anhydrous DMF (10 mL) and stirred at room temperature overnight. The mixture was poured into ice-cold water, and the resulting precipitate was filtered off, dried, and crystallized from DCM–EtOH to give compounds 8a–8n in yield of 68–80%.General procedure for the synthesis of 2-cyano-2-(4-oxo-3-arylthiazolidin-2-ylidene)acetic acid (9a–9n)
To a solution of tert-butyl acetate derivative 8 (5 mmol) in DCM (50 mL) was added a mixture of TFA (7.5 mL) and DCM (75 mL). The mixture was stirred at room temperature until the reaction was complete as indicated by TLC (typically 24 h). The solvent was evaporated under reduced pressure. The residual solid was further crystallized from DCM–MeOH to afford the compounds 9a–9n in yield of 84–91%.General procedure for the synthesis of ATs (10a–10n)
Carboxylic acid compound 9 (0.5 mmol) was added to a solution of NaBH4 (1 mmol) in water (2.5 mL) at 15 °C. The reaction mixture was stirred at 15 °C until the reaction was complete as determined by TLC analysis (typically 0.5–24 h). The reaction mixture was quenched with 1 M HCl, and the product precipitated was filtered. The solid was purified by silica gel column chromatography (PE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EA = 5:1) to afford the compounds 10a–10n.
:EA = 5:1) to afford the compounds 10a–10n.
General procedure for the synthesis of ADHTs (11a–11m)
Carboxylic acid compound 9 (0.5 mmol) was added to a solution of KBH4 (5 mmol) in water (5 mL) at 60 °C. The reaction mixture was stirred at 60 °C for 0.5 h. The reaction mixture was cooled to room temperature and quenched with 1 M HCl. The aqueous layer was extracted with EtOAc (3 × 2 mL). The combined organic phases were washed once with brine, dried with anhydrous Na2SO4, vacuum-filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (PE:EA = 3:1) to afford the corresponding ADHTs 11a–11m.
Acknowledgements
This work was financial supported by Shanghai Foundation of Science and Technology (15431902100) and National Natural Science Foundation of China (21272071).References
- D. Gramec, L. Peterlin Mašič and M. Sollner Dolenc, Chem. Res. Toxicol., 2014, 27, 1344 Search PubMed.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832 CrossRef CAS PubMed.
- (a) S. Arrechea, J. N. Clifford, L. Pellejà, A. Aljarilla, P. de la Cruz, E. Palomares and F. Langa, Dyes Pigm., 2016, 126, 147 CrossRef CAS; (b) Y. Cao, Y. Bai, Q. Yu, Y. Cheng, S. Liu, D. Shi, F. Gao and P. Wang, J. Phys. Chem. C, 2009, 113, 6290 CrossRef CAS; (c) K. Hara, M. Kurashige, Y. Dan-oh, C. Kasada, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, New J. Chem., 2003, 27, 783 RSC; (d) Z.-S. Wang, Y. Cui, Y. Dan-oh, C. Kasada, A. Shinpo and K. Hara, J. Phys. Chem. C, 2007, 111, 7224 CrossRef CAS.
- (a) G. Barbarella, M. Melucci and G. Sotgiu, Adv. Mater., 2005, 17, 1581 CrossRef CAS; (b) T. Krishnamoorthy, F. Kunwu, P. P. Boix, H. Li, T. M. Koh, W. L. Leong, S. Powar, A. Grimsdale, M. Grätzel and N. Mathews, J. Mater. Chem. A, 2014, 2, 6305 RSC; (c) Y. A. Udum, E. Yildiz, G. Gunbas and L. Toppare, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3723 CrossRef CAS.
- (a) M. Halik, H. Klauk, U. Zschieschang, G. Schmid, S. Ponomarenko, S. Kirchmeyer and W. Weber, Adv. Mater., 2003, 15, 917 CrossRef CAS; (b) C. Rost, S. Karg, W. Riess, M. A. Loi, M. Murgia and M. Muccini, Appl. Phys. Lett., 2004, 85, 1613 CrossRef CAS.
- K. Doré, S. Dubus, H.-A. Ho, I. Lévesque, M. Brunette, G. Corbeil, M. Boissinot, G. Boivin, M. G. Bergeron and D. Boudreau, J. Am. Chem. Soc., 2004, 126, 4240 CrossRef PubMed.
- (a) D. M. Vriezema, J. Hoogboom, K. Velonia, K. Takazawa, P. Christianen, J. Maan, A. E. Rowan and R. J. Nolte, Angew. Chem., 2003, 115, 796 CrossRef; (b) H. Yu, A. E. Pullen, M. G. Büschel and T. M. Swager, Angew. Chem., Int. Ed., 2004, 43, 3700 CrossRef CAS PubMed.
- (a) S. Dong and L. A. Paquette, J. Org. Chem., 2005, 70, 1580 CrossRef CAS PubMed; (b) D. H. Martyres, J. E. Baldwin, R. M. Adlington, V. Lee, M. R. Probert and D. J. Watkin, Tetrahedron, 2001, 57, 4999 CrossRef CAS; (c) J. Renault, D. Laduree and M. Robba, Nucleosides Nucleotides, 1994, 13, 1135 CrossRef CAS; (d) M.-G. A. Shvekhgeimer, Chem. Heterocycl. Compd., 1998, 34, 1101 CrossRef CAS; (e) W. Zhu, Y. Chong, H. Choo, J. Mathews, R. F. Schinazi and C. K. Chu, J. Med. Chem., 2004, 47, 1631 CrossRef CAS PubMed; (f) S. Benetti, C. De Risi, G. P. Pollini and V. Zanirato, Chem. Rev., 2012, 112, 2129 CrossRef CAS PubMed; (g) J. Sun, L.-L. Zhang, E.-Y. Xia and C.-G. Yan, J. Org. Chem., 2009, 74, 3398 CrossRef CAS PubMed.
- E. S. Darwish, Molecules, 2008, 13, 1066 CrossRef CAS PubMed.
- H. Maruoka, M. Yamazaki and Y. Tomioka, J. Heterocycl. Chem., 2004, 41, 641 CrossRef CAS.
- H. Maruoka, K. Yamagata and M. Yamazaki, J. Heterocycl. Chem., 2001, 38, 269 CrossRef CAS.
- (a) Q. Huang, P. F. Richardson, N. W. Sach, J. Zhu, K. K.-C. Liu, G. L. Smith and D. M. Bowles, Org. Process Res. Dev., 2011, 15, 556 CrossRef CAS; (b) A. M. Shestopalov, L. A. Rodinovskaya and A. A. Shestopalov, J. Comb. Chem., 2009, 12, 9 CrossRef PubMed; (c) G. Sommen, A. Comel and G. Kirsch, Synthesis, 2003, 735 CAS; (d) G. Sommen, A. Comel and G. Kirsch, Synlett, 2001, 1731 CrossRef CAS; (e) G. Sommen, A. Comel and G. Kirsch, Tetrahedron Lett., 2002, 43, 257 CrossRef CAS; (f) L. Luo, L. Meng, Q. Sun, Z. Ge and R. Li, Tetrahedron Lett., 2014, 55, 259 CrossRef CAS; (g) G. Sommen, A. Comel and G. Kirsch, Tetrahedron, 2003, 59, 1557 CrossRef CAS; (h) A. El-Shafei, H. Abdel-Ghany, A. Sultan and A. El-Saghier, Phosphorus, Sulfur Silicon Relat. Elem., 1992, 73, 15 CrossRef CAS.
- O. G. A. Tarasova, L. V. Klyba, V. Y. Vvedensky, N. A. Nedolya, B. A. Trofimov, L. Brandsma and H. D. Verkruijsse, Eur. J. Org. Chem., 1998, 253 CrossRef CAS.
- A. R. Katritzky, X. Wang and A. Denisenko, J. Org. Chem., 2001, 66, 2850 CrossRef CAS PubMed.
- M. M. M. Raposo, A. M. Sampaio and G. Kirsch, Synthesis, 2005, 199 CrossRef CAS.
- D. Virieux, A. F. Guillouzic and H. J. Cristau, Heteroat. Chem., 2007, 18, 312 CrossRef CAS.
- K. H. Kim, J. W. Lim, S. Y. Kim and J. N. Kim, Tetrahedron Lett., 2015, 56, 5799 CrossRef CAS.
- V. Dotsenko, S. Krivokolysko, A. Chernega and V. Litvinov, Russ. Chem. Bull., 2007, 56, 1431 CrossRef CAS.
- (a) A. V. Samet, A. M. Shestopalov, V. N. Nesterov and V. V. Semenov, Synthesis, 1997, 623 CrossRef CAS; (b) A. Shestopalov, O. Bogomolova and V. Litvinov, Synthesis, 1991, 277 CrossRef CAS.
- P. Gopinath and S. Chandrasekaran, J. Org. Chem., 2011, 76, 700 CrossRef CAS PubMed.
- (a) F. C. Brown, Chem. Rev., 1961, 61, 463 CrossRef CAS; (b) M. Farhat, A. El-Saghier, M. Makhlouf, K. Kreddan and A. Elmezoughi, J. Sulfur Chem., 2007, 28, 563 CrossRef CAS; (c) M. F. Farhat, M. A. Makhlouf, A. M. El-Saghier, A. B. Mezoughi, S. M. Awhida and A. A. El-mehdi, Arabian J. Chem., 2011, 4, 307 CrossRef CAS; (d) M. M. Hanna and R. F. George, Chem. Pharm. Bull., 2012, 60, 1195 CrossRef CAS PubMed; (e) D. Kaminskyy, A. K. Gzella and R. Lesyk, Synth. Commun., 2014, 44, 231 CrossRef CAS; (f) G. R. Newkome and A. Nayak, Adv. Heterocycl. Chem., 1980, 25, 83 CrossRef; (g) H. M. Refaat, Eur. J. Med. Chem., 2010, 45, 2949 CrossRef CAS PubMed; (h) A. C. Tripathi, S. J. Gupta, G. N. Fatima, P. K. Sonar, A. Verma and S. K. Saraf, Eur. J. Med. Chem., 2014, 72, 52 CrossRef CAS PubMed; (i) A. Verma and S. K. Saraf, Eur. J. Med. Chem., 2008, 43, 897 CrossRef CAS PubMed.
- (a) A. B. Prasad, J. B. Kanth and M. Periasamy, Tetrahedron, 1992, 48, 4623 CrossRef CAS; (b) R. Tale, K. Patil and S. Dapurkar, Tetrahedron Lett., 2003, 44, 3427 CrossRef CAS; (c) R. P. McGeary, Tetrahedron Lett., 1998, 39, 3319 CrossRef CAS; (d) R. Sharma, G. H. Voynov, T. V. Ovaska and V. E. Marquez, Synlett, 1995, 839 CrossRef CAS; (e) J. B. Kanth and M. Periasamy, J. Org. Chem., 1991, 56, 5964 CrossRef CAS.
- (a) G. L. Khatik, A. Pal, S. M. Mobin and V. A. Nair, Tetrahedron Lett., 2010, 51, 3654 CrossRef CAS; (b) P. Liu, C. Li, J. Zhang and X. Xu, Synth. Commun., 2013, 43, 3342 CrossRef CAS.
- (a) L. Citrome, Int. J. Clin. Pract., 2009, 63, 140 CrossRef CAS PubMed; (b) A. Leyva-Pérez, J. R. Cabrero-Antonino and A. Corma, Tetrahedron, 2010, 66, 8203 CrossRef.
- (a) A. K. Gzella, M. Kowiel, A. Suseł, M. N. Wojtyra and R. Lesyk, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 70, 812 CrossRef CAS PubMed; (b) H. H. Abdu-Allah, S. G. Abdel-Moty, R. El-Awady and A.-N. A. El-Shorbagi, Bioorg. Med. Chem. Lett., 2016, 26, 1647 CrossRef CAS PubMed; (c) J. Váňa, J. Hanusek, A. Růžička and M. Sedlák, J. Heterocycl. Chem., 2009, 46, 635 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1476608 and 1476609. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra11151c |
| This journal is © The Royal Society of Chemistry 2016 |