
TEMPO-catalyzed synthesis of 5-substituted isoxazoles from propargylic ketones and TMSN3†
Yan He‡
a,
Yu-yang Xie‡a,
Ying-chun Wangb,
Xiao-min Bina,
Da-chao Hua,
Heng-shan Wang*a and
Ying-ming Pan*a
aState Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources, School of Chemistry and Pharmaceutical Sciences of Guangxi Normal University, Guilin 541004, People's Republic of China. E-mail: whengshan@163.com; panym2013@hotmail.com
bCollege of Chemistry and Chemical Engineering, Jishou University, Jishou 416000, People's Republic of China
First published on 13th June 2016
Abstract
A novel and efficient TEMPO-catalyzed synthesis of 5-substituted isoxazoles from propargylic ketones and TMSN3 via a radical mechanism process is described. This methodology provides an easy access to a variety of useful 5-substituted isoxazoles from simple and readily available propargylic ketones and TMSN3 in good to excellent yields. A plausible reaction mechanism for this process is proposed.
Introduction
The isoxazole core, one of the important five-membered nitrogen heterocycles, occupies an important place in organic chemistry1 because of its wide applications in medicinal chemistry,2 materials science,3 biologically active molecules,4 and as an intermediate in organic synthesis.5 For this reason, considerable research efforts have been focused on the development of novel and efficient methods for the synthesis of isoxazoles. There are three main methodologies: the cyclization of ketoxime dianions or propargylic oximes (Scheme 1A),6 [3 + 2] cycloaddition reactions (Scheme 1B),7 and condensations with hydroxylamine (Scheme 1C).8 For example, in 2005, Larock and co-workers reported the isoxazole products obtained from cycloisomerizations of propargylic oximes with ICl, I2, Br2, or PhSeBr.6a The Fokin group in 2008 reported a Ru(II)-catalyzed [3 + 2] cycloaddition of alkynes with nitrile oxides to give 3,4-disubstituted isoxazoles.7b And in 2014, Liang reported the synthesis of disubstituted isoxazoles from homopropargylic alcohol, t-BuONO, and H2O via condensation with hydroxylamine.8c Very recently, Reddy et al. reported the direct tandem azidation and denitrogenative cyclization of internal propargylic ketones with trimethylsilyl azide as an amino surrogate (Scheme 1D).9 However, they did not elaborate their methodology with unsubstituted propargylic ketones. Although, the poly-substituted isoxazoles could be get by these methods which are general, regioselective, and high yielding, only a limited number of methods for the synthesis of 5-substituted isoxazoles have been described.10 Furthermore, some of these methods face the limitation of low atom efficiency, substrate specificity or forcing conditions. Therefore, the development of new methods for the regioselective synthesis of 5-substituted isoxazoles under mild reaction conditions is still highly desired. | ||
| Scheme 1 Synthesis of isoxazoles. | ||
Recently, organocatalysis has attracted considerable attention and has been significantly developed.11 2,2,6,6-Tetramethylpiperidine 1-oxyl (TEMPO), a shelf-stable radical species, is the catalyst of choice in industry and has been widely used for the oxidation of alcohols to carbonyls.12 Notably, TEMPO has been reported as a nonmetal catalyst in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double-bond cleavage to produce oxo nitriles.13 Herein, we report a TEMPO catalyzed unsubstituted propargylic ketones to produce 5-substituted isoxazoles using a useful reagent TMSN3 as the nitrogen source (Scheme 1).
C double-bond cleavage to produce oxo nitriles.13 Herein, we report a TEMPO catalyzed unsubstituted propargylic ketones to produce 5-substituted isoxazoles using a useful reagent TMSN3 as the nitrogen source (Scheme 1).
Results and discussion
At the outset of our studies, the reaction of propargylic ketone 1a with TMSN3 was attempted under different conditions (Table 1). When the reaction was catalyzed by TEMPO, no desired product was detected with most of propargylic ketone 1a remained (Table 1, entry 1). Then, we were glad to find that propargylic ketone 1a was converted into isoxazole 2a when Mn(OAc)2·2H2O (15 mol%) was employed (Table 1, entry 2). We conjectured that the reaction were carried out in the presence of H2O. Therefore, various catalyst were subsequently screened in this reaction, such as CuBr, MnO2, MnCl2, MnBr2 and TEMPO (Table 1, entries 3–7). Among them, TEMPO was found to be the optimal one and afforded product 2a in 76% yield (Table 1, entry 7). Subsequently, various solvents examined, DMSO, toluene, CH2Cl2, 1,4-dioxane and DMF turned out to give the product (Table 1, entries 8–12). To our delight, in CH3OH without H2O, TEMPO performed well with high effectivity delivering the product in good yield (78% isolated yield, Table 1, entry 13). CH2Cl2 and 1,4-dioxane without H2O were tested, we can not get the desired product (Table 1, entries 14–15).
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.3 mmol), TMSN3 (0.45 mmol), catalyst (15 mol%) in solvent (2 mL) at room temperature for 12 h under air. PPh3 (1.0 equiv.) was added after 12 h and the mixture was stirred for another 1 h.b Isolated yield of pure product based on 1a.c 0.2 mL H2O was added. | |||
| 1 | TEMPO | CH3CN | N.R. |
| 2 | Mn(OAc)2·2H2O | CH3CN | 55% |
| 3c | CuBr | CH3CN/H2O | N.R. |
| 4c | MnO2 | CH3CN/H2O | N.R. |
| 5c | MnCl2 | CH3CN/H2O | N.R. |
| 6c | MnBr2 | CH3CN/H2O | 63% |
| 7c | TEMPO | CH3CN/H2O | 76% |
| 8c | TEMPO | DMSO/H2O | 40% |
| 9c | TEMPO | Toluene/H2O | 47% |
| 10c | TEMPO | CH2Cl2/H2O | 71% |
| 11c | TEMPO | 1,4-Dioxane/H2O | 74% |
| 12 | TEMPO | DMF/H2O | 69% |
| 13 | TEMPO | CH3OH | 78% |
| 14 | TEMPO | CH2Cl2 | N.R. |
| 15 | TEMPO | 1,4-Dioxane | N.R. |

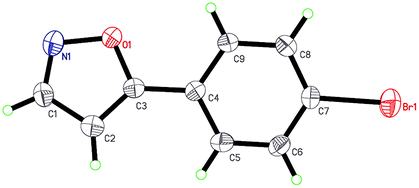
With the optimized conditions in hand, the scope of substrates that could participate in the reaction was next investigated (Table 2). The acetylenic ketones 1a–1s were either commercially available or prepared using a known procedure (see ESI†). To our delight, aryl–alkynyl ketones bearing various substitutes participated well in the cyclization reaction, gave 5-substituted isoxazoles 2a–2j in moderate to good yields (70–85%). And the substrate with an electron-withdrawing group on the aromatic ring gave better yield than that of an electron-donating group on the aromatic ring. The crystallization of compound 2g from ethanol gave a single crystal suitable for X-ray analysis. Fig. 1 illustrates the molecular structure of the 5-substituted isoxazole 2g. When the aryl group was substituted in the 2-, 3-, and 4-positions by a CH3 group, the corresponding isoxazole compound could be synthesized in 76%, 78%, and 77% yield, respectively. Gratifyingly, 5-(2-bromo-5-methoxyphenyl)isoxazole (1i) and 5-(4-(tert-butyl)phenyl)isoxazole (1j) were found to the reaction, providing the desired products 2i and 2j in 79% and 70% yield, respectively. Fortunately, propargylic ketones containing heteroaryl units or condensed rings were also tolerated (2k, 2l and 2m). It should be noted that the reactions with the alkyl acetylenic ketone, leading to moderate yields of products (2n, 2o). Similarly, propargylic ketones having a vinylic substituent (1p) and an acetylenic substituent (1q) gave the corresponding isoxazoles 2p and 2q in 67% and 66% yield, respectively. In addition, 4-phenylpent-1-yn-3-one (1r) was not an appropriate starting material under these reaction conditions. When the optically active acetylenic ketone (R)-1-(2,2′-diisopropoxy-[1,1′-binaphthalen]-3-yl)prop-2-yn-1-one was examined as a substrate, to our delight, (R)-5-(2,2′-diisopropoxy-[1,1′-binaphthalen]-3-yl)isoxazole 2s was formed in 55% yield.
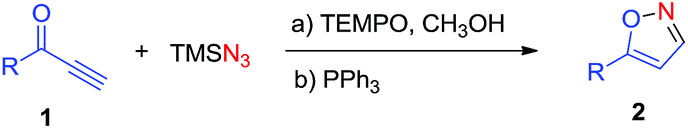

To our disappointment, internal propargylic ketones could not provide the desired product using the current catalytic system. Internal propargylic ketone 3 was tested under the standard conditions, providing the 4,5-disubstituted 1,2,3-triazole 4 in 82% yield (eqn (1)).
Some control experiments were carried out in order to explore the possible reaction pathway. CH318OH and 18O2 isotopic labeling experiments were investigated. As expected, the oxygen atom of 2a originated from propargylic ketone 1a (eqn (2) and (3), the results were determined by HRMS, see ESI†). Furthermore, CH3OD was investigated to the reaction and produced the deuterated product 2a–d in 78% yield (eqn (4)). And the deuterated substrate 1a–d′ was subjected to the reaction and produced the product 5-phenylisoxazole 2a–d′ in 77% yield (eqn (5)).
On the basis of the preliminary results, a plausible pathway is proposed for this cascade reaction (Scheme 2). Initially, TMSN3 generates azido free radical associated with the formation of intermediate A. The TEMPO–TMS species A reacts with CH3OH to form TEMPOH B and CH3O–TMS. The azido free radical subsequently attacks the alkyne to form radical intermediate 5. Subsequently, the radical 5 reacts directly with the TEMPOH species B, yields the intermediate 6 which is detected by HRMS (see ESI†), with the regeneration of the catalyst. The intramolecular azide–alkene cycloaddition was performed to afford triazole species 7. This triazole 7 undergoes homolytic cleavage to give radical intermediate 8, which release N2 to give the imine 9. Then, one-electron shift, followed by a subsequently intramolecular radical coupling to afford the product 2a. The intermolecular [3 + 2] cycloaddition reaction of internal propargylic ketone with TMSN3 were occurred during the standard conditions because of the effect of steric hindrance.
 | ||
| Scheme 2 A plausible reaction mechanism. | ||
Conclusions
In summary, we have demonstrated a facile transformation of propargylic ketones into 5-substituted isoxazoles catalyzed by TEMPO. The reaction has been delineated to proceed by a radical mechanism. Readily available, cheap starting substrates, an inexpensive catalyst, metal-free mild reaction conditions, a simple experimental procedure, and good yields, are some of the attractive attributes of the present protocol. Further work concerning synthetic applications and biological assessment of these isoxazoles is underway and the results shall be reported in due course.Experimental
General remarks
Melting points were measured with a SGW X-4 melting point instrument (uncorrected). Proton nuclear magnetic resonance spectra (1H NMR) and carbon nuclear magnetic resonance spectra (13C NMR) were recorded at 400 MHz and 100 MHz, respectively, using CDCl3 as reference standard (δ 7.26 ppm) for 1H NMR and (δ 77 ppm) for 13C NMR. HRMS (ion trap) were recorded using ESI. Precoated silica gel plates GF-254 were used for thin-layer analytical chromatography. Column chromatography was performed on silica gel (300–400 mesh). Starting materials azidomethyl aromatics were readily prepared according to literature procedures. Unless otherwise noted, all reagents were obtained commercially and used without further purification.General procedure for the synthesis of 5-substituted isoxazoles
To the mixture of propargylic ketones (1.0 equiv., 0.3 mmol) and azides (1.5 equiv., 0.45 mmol), TEMPO (15 mol%), and 2.0 mL CH3OH at 25 °C for 12 h in the air. PPh3 (1.0 equiv.) was added after 12 h and the mixture was stirred for another 1 h. The progress of the reaction was monitored by thin-layer chromatography. Upon completion, the mixture was evaporated under reduced pressure, and the residue was separated by column chromatography (ethyl acetate/petroleum ether = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 to 1:20) to give the pure product.
:40 to 1:20) to give the pure product.
Acknowledgements
We would like to thank National Natural Science Foundation of China (21362002 and 81260472), State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources (CMEMR2014-A02 and CMEMR2012-A20), Guangxi's Medicine Talented Persons Small Highland Foundation (1306), and The Fund of Guangxi Key Laboratory of Functional Phytochemicals Research and Utilization (FPRU2015-2) for financial support.Notes and references
- (a) A. M. S. Silva, A. C. Tome, T. M. V. Pinho e Melo and J. Elguero, in Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. J. Vaquero and J. Barluenga, Wiley-VCH, Weinheim, 2011, pp. 727–808 Search PubMed; (b) B. J. Wakefield, in Science of Synthesis: HoubenWeyl Methods of Molecular Transformations, ed. E. Schaumann, Georg Thieme Verlag, Stuttgart, New York, 2004, vol. 11, pp. 229–288 Search PubMed; (c) T. M. V. D. Pinho e Melo, Curr. Org. Chem., 2005, 9, 925 CrossRef CAS.
- (a) M. Eda, T. Kuroda, S. Kaneko, Y. Aoki, M. Yamashita, C. Okumura, Y. Ikeda, T. Ohbora, M. Sakaue, N. Koyama and K. Aritomo, J. Med. Chem., 2015, 58, 4918 CrossRef CAS PubMed; (b) N. Ye, Y.-m. Zhu, H.-j. Chen, Z.-q. Liu, F.-C. Mei, C. Wild, H.-y. Chen, X.-d. Cheng and J. Zhou, J. Med. Chem., 2015, 58, 6033 CrossRef CAS PubMed; (c) M. Koufaki, A. Tsatsaroni, X. Alexi, H. Guerrand, S. Zerva and M. N. Alexis, Bioorg. Med. Chem., 2011, 19, 4841 CrossRef CAS PubMed; (d) D. A. Patrick, S. A. Bakunov, S. M. Bakunova, E. V. K. S. Kumar, R. J. Lombardy, S. K. Jones, A. S. Bridges, O. Zhirnov, J. E. Hall, T. Wenzler, R. Brun and R. R. Tidwell, J. Med. Chem., 2007, 50, 2468–2485 CrossRef CAS PubMed.
- (a) A. D. Burrows, C. G. Frost, M. F. Mahon, P. R. Raithby, C. Richardson and A. J. Stevenson, Chem. Commun., 2010, 46, 5064 RSC; (b) Y. Lee, Y. Koyama, M. Yonekawa and T. Tanaka, Macromolecules, 2009, 42, 7709 CrossRef CAS.
- (a) D. A. Hay, O. Fedorov, S. Martin, D. C. Singleton, C. Tallant, C. Wells, S. Picaud, M. Philpott, O. P. Monteiro, C. M. Rogers, S. J. Conway, T. P. C. Tumber, A. Rooney, C. Yapp, P. Filippakopoulos, M. E. Bunnage, S. Mîller, S. Knapp, C. J. Schofield and P. E. Brennan, J. Am. Chem. Soc., 2014, 136, 9308 CrossRef CAS PubMed; (b) R. M. Kumbhare, U. B. Kosurkar, M. Janaki Ramaiah, T. L. Dadmal, S. N. Pushpavalli and M. Pal-Bhadra, Bioorg. Med. Chem. Lett., 2012, 22, 5424 CrossRef CAS PubMed; (c) P. Ratcliffe, J. Maclean, L. Abernnethy, T. Clatkson, M. Dempster, A. M. Easson, D. Edwards, K. Everett, H. Feilden, P. Littlewood, D. McArthur, D. McGregor, H. MeLuskey, O. Nimz, L. A. Nisbet, R. Palin, H. Tracey and G. Walker, Bioorg. Med. Chem. Lett., 2011, 21, 2559 CrossRef CAS PubMed; (d) J. Liu, L.-F. Yu, J. B. Eaton, B. Caldarone, K. Cavino, C. Ruiz, M. Terry, A. Fedolak, D. Wang, A. Ghavami, D. A. Lowe, D. Brunner, R. J. Lukas and A. P. Kozikowski, J. Med. Chem., 2011, 54, 7280 CrossRef CAS PubMed.
- (a) P. J. Lindsay-Scott, A. Clarke and J. Richardson, Org. Lett., 2015, 17, 476 CrossRef CAS PubMed; (b) R. C. F. Jones, A. Chatterley, R. Marty, W. M. Owtonb and M. R. J. Elsegood, Chem. Commun., 2015, 51, 1112 RSC; (c) P. A. Allegretti and E. M. Ferreira, Chem. Sci., 2013, 4, 1053 RSC; (d) B. Heasley, Angew. Chem., Int. Ed., 2011, 50, 8474 CrossRef CAS PubMed; (e) V. Singh, R. Saxena and S. Batra, J. Org. Chem., 2005, 70, 353 CrossRef CAS PubMed.
- (a) J. P. Waldo and R. C. Larock, Org. Lett., 2005, 7, 5203 CrossRef CAS PubMed; (b) J. P. Waldo and R. C. Larock, J. Org. Chem., 2007, 72, 9643 CrossRef CAS PubMed; (c) Z. She, D. Niu, L. Chen, M. A. Gunawan, X. Shanja, W. H. Hersh and Y. Chen, J. Org. Chem., 2012, 77, 3627 CrossRef CAS PubMed; (d) M. Ueda, S. Sugita, A. Sato, T. Miyoshi and O. Miyata, J. Org. Chem., 2012, 77, 9344 CrossRef CAS PubMed; (e) A. Sperança, B. Godoi and G. Zeni, J. Org. Chem., 2013, 78, 1630 CrossRef PubMed; (f) Y. Jeong, B. I. Kim, J. K. Lee and J. S. Ryu, J. Org. Chem., 2014, 79, 6444 CrossRef CAS PubMed; (g) W. Chen, B. Wang, N. Liu, D. Huang, X. Wang and Y. Hu, Org. Lett., 2014, 16, 6140 CrossRef CAS PubMed.
- (a) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef CAS PubMed; (b) S. Grecian and V. V. Fokin, Angew. Chem., Int. Ed., 2008, 47, 8285 CrossRef CAS PubMed; (c) H. Zheng, R. McDonald and D. G Hall, Chem.–Eur. J., 2010, 16, 5454 CrossRef CAS PubMed; (d) A. M. Jawalekar, E. Reubsaet, F. P. J. T. Rutjes and F. L. V. Delft, Chem. Commun., 2011, 47, 3198 RSC; (e) D. C. Schmitt, L. Lam and J. S. Johnson, Org. Lett., 2011, 13, 5136 CrossRef CAS PubMed; (f) W. Chen, J. Zhang, B. Wang, Z. Zhao, X. Wang and Y. Hu, J. Org. Chem., 2015, 80, 2413 CrossRef CAS PubMed; (g) K. C. Coffman, T. A. Palazzo, T. P. Hartley, J. C. Fettinger, D. J. Tantillo and M. J. Kurth, Org. Lett., 2013, 15, 2062 CrossRef CAS PubMed; (h) M. Hu, X. He, Z. Niu, Z. Yan, F. Zhou and Y. Shang, Synthesis, 2014, 46, 510 CrossRef.
- (a) S. Tang, J. He, Y. Sun, L. He and X. She, J. Org. Chem., 2010, 75, 1961 CrossRef CAS PubMed; (b) D. Xiang, X. Xin, X. Liu, R. Zhang, J. Yang and D. Dong, Org. Lett., 2012, 41, 644 CrossRef PubMed; (c) P. Gao, H. X. Li, X. H. Hao, D. P. Jin, D. Q. Chen, X. B. Yan, X. X. Wu, X. R. Song, X. Y. Liu and Y. M. Liang, Org. Lett., 2014, 16, 6298 CrossRef CAS PubMed; (d) R. Harigae, K. Moriyama and H. Togo, J. Org. Chem., 2014, 79, 2049 CrossRef CAS PubMed.
- G. R. Kumar, Y. K. Kumar and M. S. Reddy, Chem. Commun., 2016, 52, 6589 RSC.
- (a) P. Passacantilli, S. Pepe, G. Piancatelli, D. Pigini and A. Squarcia, Tetrahedron, 2004, 60, 6453 CrossRef CAS; (b) H. G. Weinig, P. Passacantilli, M. Colapietro and G. Piancatelli, Tetrahedron Lett., 2002, 43, 4613 CrossRef CAS; (c) F. A. Rosa, P. Machado, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, J. Heterocycl. Chem., 2008, 45, 879 CrossRef CAS; (d) R. M. Adlington, J. E. Baldwin, D. Catterick, G. J. Pritchard and L. T. Tang, J. Chem. Soc., Perkin Trans. 1, 2000, 2311 RSC; (e) D. H. Churykau and O. G. Kulinkovich, Synlett, 2006, 20, 3427 Search PubMed; (f) U. Klein and W. Steglich, Liebigs Ann. Chem., 1989, 247 CrossRef CAS.
- (a) L. S. Aitken, N. R. Arezki, A. Dell'Isola and A. J. A. Cobb, Synthesis, 2013, 45, 2627 CrossRef CAS; (b) A. S. Kucherenko, D. E. Siyutkin, O. V. Maltsev, S. V. Kochetkov and S. G. Zlotin, Russ. Chem. Bull., 2012, 61, 1313 CrossRef CAS; (c) B. Bradshaw and J. Bonjoch, Synlett, 2012, 23, 337 CrossRef CAS; (d) A. Łukasz, J. Hao and A. J. Karl, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef PubMed; (e) M. Benaglia and S. Rossi, Org. Biomol. Chem., 2010, 8, 3824 RSC.
- (a) P. Galletti, M. Pori, F. Funiciello, R. Soldati, A. Ballardini and D. Giacomini, ChemSusChem, 2014, 7, 2684 CrossRef CAS PubMed; (b) N. Tamura, T. Aoyama, T. Takido and M. Kodomari, Synlett, 2012, 23, 1397 CrossRef CAS; (c) T. Ludger and S. Armido, Angew. Chem., Int. Ed., 2011, 50, 5034 CrossRef PubMed; (d) X. Q. Li and C. Zhang, Synthesis, 2009, 1163 CAS.
- T. Wang and N. Jiao, J. Am. Chem. Soc., 2013, 135, 11692 CrossRef CAS PubMed.
- K. Bowden and E. R. H. Jones, J. Chem. Soc., 1946, 953 RSC.
- Y. I. Lin and S. A. Lang Jr, J. Org. Chem., 1980, 45, 4857 CrossRef CAS.
- F. A. Rosa, P. Machado, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, J. Heterocycl. Chem., 2008, 45, 879 CrossRef CAS.
- E. V. Vashkevich, V. I. Potkin and N. G. Kozlov, Russ. J. Org. Chem., 2004, 40, 1503 CrossRef CAS.
- L. Panizzi and M. Elios, Gazz. Chim. Ital., 1947, 77, 556 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental procedures, and spectral data, NMR spectra, high resolution mass spectra for all compounds, and X-ray crystallographic files (CIF) for 2g. CCDC 1449503. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra11099a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |