
Mechanical and thermal properties of toluene diisocyanate-modified cellulose nanocrystal nanocomposites using semi-crystalline poly(lactic acid) as a base matrix†
Abstract
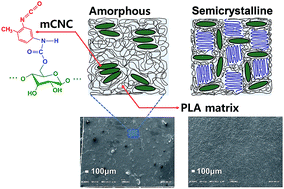
Although chemical modifications (grafting ‘onto’) of CNCs have been successfully adopted to enhance their dispersibility in apolar matrices and solvents, the problem of the dispersion level of mCNCs (chemically modified CNCs) in apolar matrices above a certain loading of nanoparticles remains an issue. CNCs were successfully modified using toluene diisocyanate, and the effects of the molar mass (Mw) and crystallinity (Xc) of semicrystalline poly(lactic acid) (PLA) on the mechanical and thermal properties of mCNC filled PLA nanocomposites were investigated. An increase in the mechanical properties of the PLA nanocomposites with mCNCs implied that Mw and Xc of PLA can be key factors to improve the dispersion level of mCNCs. In our solvent dilute polymer system, despite a reduction in the crystallinity of PLA with increasing mCNC loading level, the melting temperature of the PLAs remained constant due to the mCNC effect, which hinders the chain mobility of the PLAs. The results demonstrated that a fundamental understanding of the crystallinity and molar mass of polymers as well as surface modification of CNCs can be a reasonable approach to take full advantage of the potential usage of CNCs as reinforcements.
 Please wait while we load your content...
Please wait while we load your content...

