
Computational scrutiny of the effect of N-terminal proline and residue stereochemistry in the nucleation of α-helix fold
 †*a
†*a
Abstract
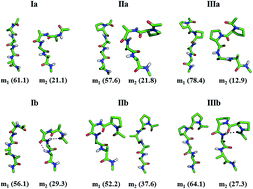
The biasing of proteins as ordered folds specific to their polypeptide sequences remains unknown in its basis. Several studies of unfolded states in folding–unfolding equilibrium with oligoalanine models have established that the polypeptide structure will unfold as ensembles that largely sample PPII and β-conformations and that the effects internal to the main chain and that of the solvent envelope are critical. While unfolded to largely extended conformations, the folded proteins are a sequence-specific mixture of β and α-conformations. The specificity is unclear in its basis, and we addressed this during the past few years with statistical mechanical studies using appropriate simpler models. In the present study, oligopeptides are used as models to elucidate the effect of N-terminal modification in the sampling of the α-conformation. Specifically, equilibrium sampling of the models for the α-conformation is assessed for dependence on the force field and the effects of specific structure perturbation in the models. Thus, Ac–LAla4–NHMe (Ia), Ac–DAla–LAla3–NHMe (Ib), Ac–LPro–LAla3–NHMe (IIa), Ac–DPro–LAla3–NHMe (IIb), Ac–LPro2–LAla2–NHMe (IIIa), and Ac–DPro–LPro–LAla2–NHMe (IIIb) are compared as N-terminal alanine or proline and L- or D-residue stereochemically perturbed models. These models are equilibrated in water as an explicit solvent using molecular dynamics, and the ordering of the polypeptide to the α-conformation is tested for the effect of force fields and the specific structural perturbation. The results of molecular dynamics ensembles imply an appreciable shift in the equilibrium sampling of conformations from the β + PPII basin to the α-basin of ϕ, ψ space, including ordering of helical microstates. The results involving well calibrated force fields imply that the ensembles appearing macroscopically as PPII helices have participating microstates that occasionally sample α-basins. We observed the nucleation of an α-helical fold with an N-terminal residue in the DPPII conformation in a mixed-L,D structure. The results imply that N-terminal L- to D-residue mutation is the stronger effect that induces folding than N-terminal alanine-to-proline or dialanine-to-diproline mutation. The present study will provide better understanding about the nucleation of helical folds in short peptides and will aid in the design of novel peptides with α-helical structures.
 Please wait while we load your content...
Please wait while we load your content...

