
Enhanced efficacy and a better pharmacokinetic profile of tamoxifen employing polymeric micelles
 *a
and
*a
and
Abstract
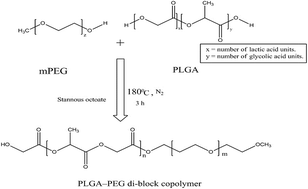
The present work aims to develop tamoxifen-loaded polymeric micelles and explore their potential in topical delivery of the drug to breast cancer cells. The PLGA–PEG copolymer was chemically synthesized and the critical micelle concentration (CMC) of the polymer was determined, drug loaded micelles were developed by a modified solvent dialysis method. The developed system was characterized for micromeritics, surface charge, drug entrapment and morphology. Furthermore, nanocarriers were evaluated for drug-release-kinetics, erythrocyte-compatibility, in vitro cytotoxicity against MCF-7 breast cancer cells and a dermal pharmacokinetic profile. The developed system was of a spherical shape with size of 76.4 ± 2.1 nm and a neutral surface charge (−4.89 mV). The system was able to offer 60.86 ± 3.21% drug entrapment. Along with drug release controlling behaviour, the cytotoxic potential of tamoxifen against MCF-7 cell lines was substantially enhanced. Dermatokinetic studies revealed better drug availability to both the epidermis and dermis than that of the plain drug. The PLGA–PEG-based micellar system offered a safer and effective option for better delivery of tamoxifen to breast cancer with immense promise of delivery across skin.
 Please wait while we load your content...
Please wait while we load your content...

