DOI:
10.1039/C6RA10731A
(Review Article)
RSC Adv., 2016,
6, 59661-59676
Recent advances in the synthesis of vinyl sulfones
Received
26th April 2016
, Accepted 13th June 2016
First published on 15th June 2016
Abstract
Development of the methodology for the preparation of vinyl sulfones is of significant interest to organic chemists. Recently, numerous useful methods have been developed, mainly including direct sulfonylation of olefins and alkynes, decarboxylative sulfonylation of α,β-unsaturated carboxylic acids and decomposition of tosylhydrazones. This review will focus on recent achievements in vinyl sulfone synthesis and the mechanisms of the reactions are also discussed.
1. Introduction
Organosulfur compounds do not only contain fundamental functional groups, such as thiol, sulfide, or disulfide units, which make them useful synthetic intermediates, but they also exist abundantly in biological systems ranging from small natural metabolites to proteins. Thus the formation of C–S bonds is one of the fundamental transformations in organic synthesis.1 In recent years, vinyl sulfones have attracted considerable interest in the area of synthetic organic chemistry, owing to their important role in serving as key structural units of many biologically active compounds2 as well as versatile building blocks for various organic transformations.3 Therefore, considerable effort has been devoted to the development of new and efficient methods for the synthesis of vinyl sulfones. Over the past three decades, various classic synthetic routes to the vinyl sulfone skeleton have been developed, such as the Knoevenagel condensations of sulfonylacetic acids with aromatic aldehydes,4 Horner–Wadsworth–Emmons reactions of sulfonyl phosphonates and carbonyl compounds,5 β-elimination of halosulfones or selenosulfones,6 and oxidation of the corresponding vinyl sulfides.7 However, most of these methods suffer from some limitations, such as inaccessible starting materials, tedious procedures, relatively harsh reaction conditions, or generation of large amounts of unwanted byproducts. Hence, the development of efficient methods for the preparation vinyl sulfone scaffolds is desirable.
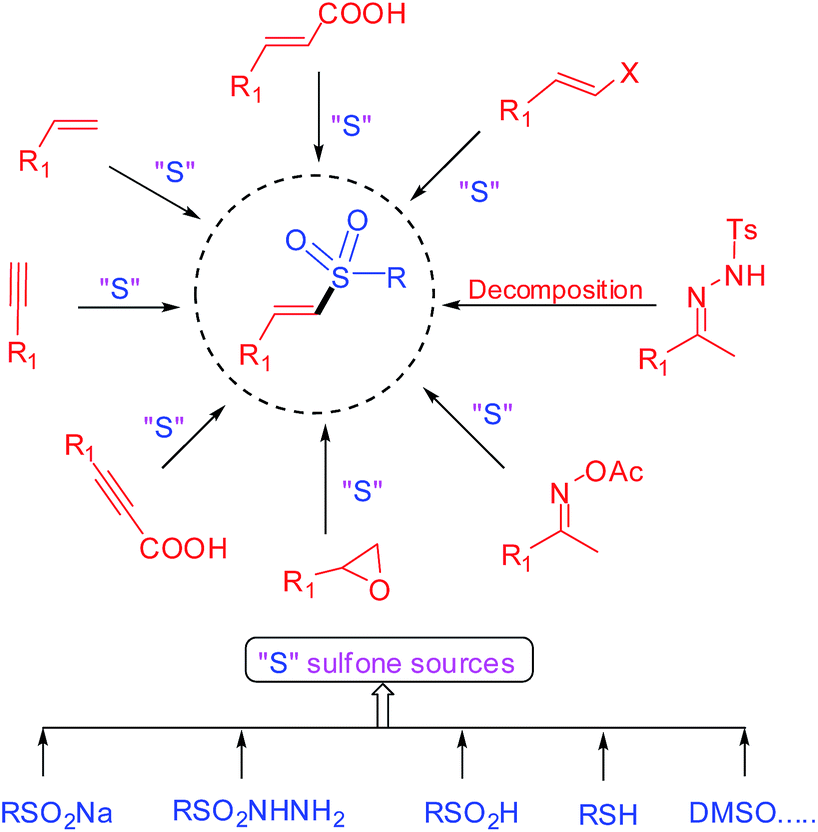
Recently, the direct cross-coupling of the sulfonyl derivative with an alkene or alkyne source has emerged rapidly as an efficient protocol for the construction of vinyl sulfones (Scheme 1). Generally, the facile sulfone sources could be originated from sodium sulfinate, sulfonyl hydrazide, sulfinic acid, thiols, DMSO and so on (Scheme 1), of which sodium sulfinate and sulfonyl hydrazide have been predominantly utilized, probably due to their good stability and ease of handling.
 |
| | Scheme 1 Recent protocols for the synthesis of vinyl sulfones. | |
The synthetic power of the vinyl sulfone functional group has been substantially upgraded over the past 30 years. Previous seminal reviews provided by Fuchs, Forristal, Gervay-Hague, Carretero, Pathak, and so on, are out-of-date.8 In most cases, the list of reviews cited in this article is an indication of the usefulness of the vinyl sulfone group and its popularity with the practising organic chemists. Thus, an updated review focused on recent achievements in vinyl sulfones synthesis as well as discussion of the mechanisms of the reactions, would enrich the arsenals of synthetic chemists who are interested in using vinyl sulfone groups in synthetic transformations. In order to restrict the topic to a manageable level, the primary organizational focus of this review will relate mainly to direct sulfonylation of olefins and alkynes, decarboxylative sulfonylation of α,β-unsaturated carboxylic acids and decomposition of tosylhydrazones in last 5 years. The cited articles were extensively selected from various journals and patents are not covered in this review. In addition, we sincerely hope that this review will serve as a handy reference for chemists interested in organic alkenyl chemistry and discovering novel and efficient methods for the preparation of vinyl sulfone building blocks.
2. Sulfonylation of alkynes
2.1 Addition of sodium sulfinates to alkynes
The use of sodium salts of sulfinic acid as sulfonylation reagents has gained much attention in recent years.9 Sodium sulfinates are stable, easy to handle and readily available from their corresponding sulfonyl chlorides. Recently, Jiang's group10 described a simple and efficient palladium-catalyzed cross-coupling of sodium sulfinates and alkynes for the synthesis of vinyl sulfones (Scheme 2). This transformation proceeds under ligand-free and mild conditions, providing excellent yields of coupling products. The results suggest that electron-poor terminal alkynes, methyl propiolate or ethyl propiolate provided the desired products in higher yields. And substituted benzenesulfinic acid sodium salts, 2-thienylsulfinic acid sodium salt, ethanesulfinic acid sodium salt or cyclopropanesulfinic acid sodium salt were successfully employed in this process, affording the desired products in good yields.
 |
| | Scheme 2 Palladium-catalyzed cross-coupling reaction of sodium sulfinates with alkynes. | |
In 2013, Kuhakarn and co-workers11 reported an improved and convenient experimental procedure to synthesize (E)-β-iodovinyl sulfones from alkynes by molecular iodine-mediated one-pot iodosulfonation (Scheme 3). The corresponding (E)-β-iodovinyl sulfones derived from phenylacetylene derivatives were obtained in acceptable yields (74–82%) while poor yield was observed with that derived from aliphatic alkyne (13%). And the reaction was believed to mechanistically proceed via radical pathway.
 |
| | Scheme 3 Molecular iodine-mediated β-iodosulfonation of sodium sulfinates with alkynes. TS, p-toluene sulfonyl. | |
Interestingly, Taniguchi12 displayed that copper-catalyzed sulfonylation of alkynes with sodium sulfinates in air afforded regio- and stereoselectively (E)-vinyl sulfones (Scheme 4). When a CuCl catalyst was employed, the hydrosulfonylation proceeded syn-selectively, and (E)-alkenyl sulfones were synthesized. In contrast, the reaction using CuI catalyst produced (E)-β-haloalkenyl sulfones anti-selectively in the presence of potassium halides. And the (E)-β-bromoalkenyl sulfones are possible to convert into various alkenyl sulfones by Suzuki–Miyaura coupling.
 |
| | Scheme 4 Plausible mechanism of copper-catalyzed sulfonylation of alkynes with sodium sulfinates. | |
Very recently, Moran and Rodríguez13 reported a facile access into otherwise difficult to obtain alkyl alkynyl sulfones and cyclic vinyl sulfones via the reaction of alkyl sulfinates with alkynyl(aryl)iodonium salts (Scheme 5). Alkynyl(aryl)iodonium salts can react with soft nucleophiles to generate alkylidene carbene intermediates under aprotic conditions (Scheme 5). These carbenes can undergo 1,2-rearrangement to form a new alkyne or insert into a C–H bond to form a five-membered ring. In this case, 1,2-rearrangement is the exclusive reaction pathway when the sulfinate contains a electron-withdrawing group.
 |
| | Scheme 5 Reaction of alkyl sulfinates with alkynyl(aryl)iodonium salts. | |
2.2 Sulfonylation of alkynes with sulfonylhydrazides
It is important to note that sulfonyl radicals generated from various sulfonyl compounds, such as sulfonyl halides,14 selenides,15 cyanides,16 azides,17 and sodium sulfinates,18 etc. Recently, Taniguchi has demonstrated that sulfonyl radicals can be generated from the corresponding hydrazine compounds.19 In 2013, Xu and co-workers20 combined sulfonylhydrazides with FeCl3 or FeBr3, which was used as the Cl and Br source, to allow chlorosulfonylation or bromosulfonylation of alkynes (Scheme 6). Generally, bromosulfonylation and chlorosulfonylation exhibited similarly reactivity, both giving moderate yields. A mechanism involving radical pathway was given (Scheme 6). Firstly, sulfonyl radicals were generated from sulfonylhydrazides under the irritation of the tert-butoxyl and tert-butylperoxy radicals. Then, addition of sulfonyl radical to the Fe-coordinated alkynes was subsequently occurred. At last, the (E)-β-halovinylsulfones were afforded through reductive elimination of Fe(IV) intermediate to the Fe(II) catalyst.
 |
| | Scheme 6 Chlorosulfonylation or bromosulfonylation of alkynes with sulfonylhydrazides. TBHP, tert-butyl hydroperoxide. | |
Interestingly, Li and co-workers21 reported an efficient and simple protocol for the preparation of (E)-β-iodovinylsulfones by TBHP mediated reaction of aryl acetylenes with sulfonylhydrazides and iodine (Scheme 7). The reactions of various substituted phenylacetylenes with TsNHNH2 were examined. Good to excellent yields were observed with halogen, trifluoromethyl, methyl, and n-pentyl substituted phenylacetylenes. Heteroaryl acetylenes, such as pyridyl and thiophenyl acetylenes were also compatible substrates and delivered the corresponding (E)-β-iodovinylsulfones. However, the yield diminished markedly when aliphatic alkyne was used. A sulfonyl radical addition to aryl acetylenes and subsequent trapping of the C-centered alkenyl radicals intermediate with I2 or iodine free radicals, was believed to be the mechanism (Scheme 7). It is also possible that the reaction might occur through addition of arenesulphonyl iodides to aryl acetylenes.
 |
| | Scheme 7 TBHP mediated reaction of sulfonylhydrazides and iodine with aryl acetylenes. | |
In 2015, a new approach to the selective synthesis of (E)-vinylsulfones via a Fe/Cu co-catalyzed sulfonylation of phenylacetylene with sulfonylhydrazides was demonstrated by Mao and co-workers (Scheme 8).22 Several alkyl and halo substituted aryl alkynes and heterocyclic alkynes, furnished the reactions smoothly affording the desired products in moderate to good yields, with exclusively E isomers obtained (Scheme 8). And the reaction was believed to mechanistically proceed via sulfonyl radical addition/reductive elimination pathway.
 |
| | Scheme 8 Fe/Cu co-catalyzed sulfonylation of phenylacetylene with sulfonylhydrazides. DTBP, di-tert-butyl peroxide. | |
Very recently, the hydrosulfonylation of terminal alkynes with sulfonylhydrazides reaction using copper(I) as the catalyst was also utilized to achieve the construction of (E)-vinyl sulfone scaffolds23 (Scheme 9). This hydrosulfonylation proceeds smoothly in moderate yields, and this transformation features a good tolerance of substrates including aliphatic sulfonylhydrazides. A plausible reaction mechanism involving sulfonyl radical is also given, which is similar to the literature reported by Mao.22
 |
| | Scheme 9 Copper-catalyzed hydrosulfonylation of terminal alkynes with sulfonylhydrazides. 2,2′-bpy, 2,2′-bipyridine. | |
2.3 Sulfonylation of alkynes with arylsulfinic acids
Sulfonyl radicals could be generated from the corresponding sulfinic acids and were involved in the procedures for the oxidative difunctionalization of alkenes and alkynes, which was disclosed recently by Lei' group.24 Under transition-metal or metal-free conditions, sulfonyl radical addition to alkynes or alkenes were both exclusively given the trans-configured β-substituted vinyl sulfones, whose behavior is similar to the anti-Markovnikov addition.25 But, there are little literature reporting the efficient synthesis of a-substituted vinyl sulfone scaffolds. And these previous methods suffer from poor a/β regioselectivity and limited substrate scope.26 In 2014, Shi and co-workers27 reported that a-substituted vinyl sulfones were generally synthesized via gold-catalyzed Markovnikov addition of arylsulfinic acids to alkynes (Scheme 10). Various aromatic alkynes, heteroaromatic alkynes and aliphatic alkynes were tested and gave the corresponding products in modest to good yields. The reaction tolerated both electron-withdrawing and electron-donating groups.
 |
| | Scheme 10 Gold-catalyzed sulfonylation of terminal alkynes with arylsulfinic acids. BrettPhos, 2-(dicyclohexylphosphino)-3,6-dimethoxy-2′,4′,6′-triisopropyl-1,1′-biphenyl. TA, 1H-benzotriazole, DCE, 1,2-dichloroethane, Tf, trifluoromethanesulfonyl. | |
In 2013, Wang and co-workers28 reported an efficient approach for the construction of vinyl sulfone scaffolds via copper-catalyzed direct sulfonylation of arylsulfinic acids with alkynes (Scheme 11). In general, aromatic or heteroaromatic alkynes, and arylsulfinic acids with electron-donating or withdrawing groups were tolerated in this process to afford the corresponding products. The proposed mechanism of the reaction was shown in Scheme 11. Firstly, sulfonyl radical could be generated from arylsulfinic acid under oxidation by CuII via the single electron transfer (SET) process. Subsequently, vinyl radical was formed via the selective addition of sulfonyl radical to alkyne, which interacted with CuI species to obtain vinyl copper(II) complexes. Finally, the vinyl sulfones were given after protonation of vinyl copper(II) complexes and regenerated Cu(II) catalyst.
 |
| | Scheme 11 Copper-catalyzed sulfonylation of alkynes with arylsulfinic acids. | |
Very recently, Wang's group29 also presented a protocol of direct difunctionalization of alkynes with arylsulfinic acids and molecular iodine for the preparing (E)-β-iodovinyl sulfones (Scheme 12). Without metal catalyst or additives, various substituted (E)-β-iodovinyl sulfones in moderate to good yields with excellent stereo- and regioselectivities were provided. And a radical process was postulated in this reaction system, which sulfonyl radical species was easily generated from arylsulfinic acids under air.
 |
| | Scheme 12 Difunctionalization of alkynes with arylsulfinic acids and molecular iodine. DME, dimethyl ether. | |
2.4 Sulfonylation of alkynes with DMSO
Dimethyl sulfoxide (DMSO) has been widely used not only as a solvent but also as a reagent in chemistry. While methyl sulfonyl radicals generated from DMSO in organic synthesis are rarely reported.30 In 2014, Loh and co-workers31 demonstrated that DMSO was activated in a new catalytic system which involves copper, oxygen and HPO(OEt)2 to afford a methyl sulfonyl radical that can functionalize alkynes for synthesis of (E)-vinyl methyl sulfones (Scheme 13). The proposed reaction mechanism is given in Scheme 13. Initially, methyl sulfonyl radical was probably generated and subsequently reacted with alkynes to form vinyl radical. After hydrogen atom transfer, only E isomer vinyl methyl sulfones was detected (Scheme 13). Very recently, Zhao and co-workers32 also developed a novel and practical CuSO4·5H2O catalyzed H-phosphonate promoted synthesis of (E)-vinyl alkylsulfones from alkynes and DMSO (Scheme 14).
 |
| | Scheme 13 Copper-catalyzed sulfonylation of alkynes with DMSO. | |
 |
| | Scheme 14 Copper-catalyzed sulfonylation of alkynes with DMSO. TFA, trifluoroacetic acid. | |
2.5 Sulfonylation of alkynes with other sulfone sources (sulfonyl chlorides, 1,2-bis(phenylsulfonyl)ethane, thiols and t-butylsulfinamide)
In 2010, Deng and Zou33 illustrated the first nucleophilic substitution reaction of organoindium with sulfonyl chlorides. This reaction of vinylindium, derived from terminal alkynes via hydroindation, with sulfonyl chlorides in the presence of Ag2O provides access to (E)-vinyl sulfones (Scheme 15). A reasonable explanation is that Ag2O seems to promote the leave of chlorine as anion, while an attack by vinylindium is at the same time made in the opposite direction, thus leading to favorable generation of vinyl sulfones (Scheme 15).
 |
| | Scheme 15 Reaction of vinylindium with sulfonyl chlorides. | |
In 2011, Li and co-workers34 reported that 1,2-bis(phenylsulfonyl)ethane can act as the sulfone resource, which generated phenylsulfonyl intermediates in situ for the conjugate addition to the electron-deficient alkynes leading to the corresponding (E)-vinyl sulfones (Scheme 16). The procedure underwent the C–S bond cleavage and conjugate addition in the Pd(OAc)2/DMEDA catalytic system. The results showed that a wide variety of 3-arylpropiolamides, and several N-substituents, either alkyl or aryl groups, took place smoothly under the standard conditions. And the reaction was believed to mechanistically proceed via insertion of Pd(0) into 1,2-bis-(phenylsulfonyl)ethane/complexation with an alkyne/regioselective additions/reductive elimination pathway.
 |
| | Scheme 16 Pd-catalyzed sulfonylation of alkynes with 1,2-bis(phenylsulfonyl)ethane (DMEDA, N′,N′-dimethylethane-1,2-diamine). | |
It is an attractive protocol for the preparation of vinyl sulfone scaffolds through direct addition–oxidation of thiols with alkynes. In 2012, Zhu and co-workers35 disclosed a metal-free, highly selective approach to (E)-vinyl sulfones from hydrothiolation of alkynes (Scheme 17). It was the first example for the construction of vinyl sulfone scaffolds in one-pot by direct addition–oxidation of thiols with alkynes. Aromatic alkynes bearing substituents such as methyl, methoxy, and fluoro proceeded smoothly to afford (E)-isomeric predominant products. The optimized conditions were also consistent with aliphatic alkynes. The plausible mechanism via addition/radical trapping/oxidation process was discussed by the authors.
 |
| | Scheme 17 Metal-free hydrothiolation of alkynes. | |
Despite the remarkable improvement in vinyl sulfone synthesis, the discovery of diverse sulfone sources is still highly desired. In 2015, Chen and Zhao'group36 disclosed that (E)-vinyl sulfone scaffolds could be obtained from t-butylsulfinamide and alkynes in the CuSO4–phosphorous acid catalytic system (Scheme 18). It is the first example of the use of t-butylsulfinamide as a sulfur source to prepare vinyl sulfones. The reaction mechanism is not fully understood. While the cleavage of the N–S bond of t-butylsulfinamide, which was catalyzed by CuSO4·5H2O-phosphorous acid catalytic system, was the key step for this transformation.
 |
| | Scheme 18 Synthesis of (E)-vinyl sulfones from alkynes and t-butylsulfinamide. | |
3. Sulfonylation of alkenes
3.1 Sulfonylation of alkenes with sodium sulfinates
During the past two decades, the versatility of hypervalent iodine reagents in organic synthesis is well recognized owing to their mild, highly selective, and environmentally benign properties.37 Currently, both iodine(V) and iodine(III) reagents are widely used in organic synthesis.38 One of the most important and commercially available iodine(III) reagents is (diacetoxyiodo)benzene [PhI(OAc)2], which is easy to handle and comparable in reactivity to heavy metal containing reagents. In 2010, an efficient protocol of synthesis of vinyl sulfones under PhI(OAc)2/KI-mediated addition of aryl sulfinates to alkenes was described by Kuhakarn and co-workers39 (Scheme 19). A variety of aliphatic alkenes, including those containing an oxygen atom, a bromine atom, an aldehyde function and a 1,3-dioxane moiety, gave the corresponding products. And the optimized reaction conditions were also proved to be suitable for a range of alkyl ester derivatives. The reaction was proposed to mechanistically proceed via β-iodosulfonylation/dehydroiodination pathway (Scheme 19).
 |
| | Scheme 19 Synthesis of (E)-vinyl sulfones from alkyenes and sodium sulfinates catalyzed by PhI(OAc)2. | |
In 2011, Das and co-workers40 synthesized vinyl sulfones by treatment of alkenes with sodium arene sulfinates using potassium iodide and sodium periodate in the presence of a catalytic amount of acetic acid at room temperature, and the applicability of both aromatic and aliphatic alkenes are the notable advantages of this method (Scheme 20). Mechanistically, NaIO4 oxidizes KI to liberate I2, which converts ArSO2Na into ArSO2I. In the presence of acetic acid, the liberation of I2 was found to be rapid. Next, the reaction of ArSO2I with an olefin produces the β-iodosulfone. Finally, spontaneous elimination of HI from this β-iodosulfone furnishes the corresponding vinyl sulfones (Scheme 20).
 |
| | Scheme 20 Synthesis of (E)-vinyl sulfones from alkyenes and sodium sulfinates catalyzed by KI and NaIO4. | |
Subsequently, Kuhakarn and co-workers11 reported an improved and convenient experimental procedure to synthesize vinyl sulfones from alkenes by molecular iodine-mediated one-pot iodosulfonation followed by base-induced dehydroiodination reaction (Scheme 21a). Very recently, a molecular iodine-mediated reaction of preparing vinyl sulfone scaffolds from sodium sulfinates and alkenes using environmentally benign water as the solvent under room temperature was also demonstrated by Wang and Yang41 (Scheme 21b).
 |
| | Scheme 21 Synthesis of (E)-vinyl sulfones from alkyenes and sodium sulfinates catalyzed by I2. | |
Electrosynthesis could effectively avoid the use of hazardous or toxic oxidants and achieve oxidation and reduction reactions in mild electron-transfer conditions, demonstrating its advantages and environment friendly characteristics. In 2015, Yuan and co-workers42 reported a convenient and efficient electrochemical route for the synthesis of vinyl sulfones from sodium sulfinates and olefins (Scheme 22). A plausible reaction mechanism for the electrochemical synthesis of vinyl sulfones was also proposed (Scheme 22). At the inert graphite anode, iodine ions are oxidized to I2 (2I− → I2 + 2e−). The sulfinate sodium salt reacts with I2 to generate sulfonyl iodide intermediate, which is decomposed to give a sulfonyl radical. Then, the addition of the sulfonyl radical to olefins affords carbon-centered radical intermediate, followed by the reaction with iodine radical or I2 to form iodo-intermediate, which undergoes HI eliminating to provide the target product.
 |
| | Scheme 22 Electrosynthesis of vinyl sulfones from olefins and sodium sulfinates. | |
Using green light, the organic dye eosin Y as photocatalyst and nitrobenzene as the terminal oxidant, König and co-workers43 recently demonstrated the visible light-mediated synthesis of vinyl sulfone scaffolds from olefins with aryl sulfinates (Scheme 23). The reaction allows the conversion of a variety of aryl sulfinates and 1,2-dihydronaphthalene. Styrene derivatives with electron-donating and electron-withdrawing substituents in the para position, and the bulky naphthalene moiety were also tolerated and afforded the products. The mechanism of the photocatalytic vinyl sulfone synthesis was proposed that the photoreaction proceeds via a radical pathway and oxidative quenching cycle (Scheme 23).
 |
| | Scheme 23 Visible light-mediated synthesis of vinyl sulfones from olefins and sodium sulfinates. PC, photocatalyst. | |
3.2 Sulfonylation of alkenes with sulfonylhydrazides
Iodine, a nonmetallic element, has diverse valence states as well as moderate redox potentials, which make it possible for serving as an efficient catalyst instead of transition metals in many organic reactions.44 Recently, Lei' group45 reported their progress in the iodide-catalysed radical alkenylation of sulfonyl hydrazides with simple alkenes, and demonstrated that iodine behaves as a metal in the alkenyl functionality recovery of carbon radicals (Scheme 24). In the initial cycle, iodide was oxidized under acidic conditions, which can promote sulfonyl radical generated from sulfonyl hydrazides through dehydrogenation procedure. Subsequently, catalytic HI elimination was proceeded via alkenylation of sulfonyl radicals from various sulfonyl hydrazides, which is similar to the β-hydride elimination of transition metals (Scheme 24).
 |
| | Scheme 24 Iodide catalyzed oxidative radical alkenylation for the synthesis of alkenyl sulfones. | |
In 2014, a methodology of preparation of vinyl sulfones by means of CuI-catalyzed aerobic oxidative N–S bond cleavage of sulfonyl hydrazides, followed by cross-coupling reaction with alkenes to construct Csp2–S bonds was reported by Jiang and co-workers46 (Scheme 25). The use of aromatic and heteroaromatic alkenes as well as naphthalene led to the cross-coupling products. However, the steric effect was critical for the reactivity. While aliphatic and aromatic sulfonyl hydrazides were shown to be suitable cross-coupling partners for this oxidative transformation. The mechanism for this copper-catalyzed aerobic oxidative cross-coupling reaction was proposed (Scheme 25). Initially, sulfonyl hydrazides were oxidatively decomposed under copper and air, with the release of N2 and H2O. Subsequently, active sulfonyl cuprate was formed, which reacts further with alkenes to afford the corresponding products. The authors also speculated that DMSO might serve as a co-oxidant in this transformation.
 |
| | Scheme 25 Copper-catalyzed oxidative sulfonations of alkenes. | |
3.3 Sulfonylation of alkenes with sulfonyl chlorides
Recently, visible-light photoredox catalysis has emerged as a powerful tool to promote useful redox transformations. In 2013, Zhang and Yu47 developed the direct sulfonation of enamides with sulfonyl chlorides to generate vinyl sulfones instead of the addition of sulfonyl chlorides to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds to generate a-chlorosulfones (Scheme 26). Direct C–H functionalizations of enamides or enecarbamates under the optimized conditions proceeded with a wide scope of substrates and remarkable selectivity to give functionalized vinyl sulfones with good to excellent yields. They envisaged that sulfonyl radical, which could be generated from sulfonyl chloride under photocatalysis conditions, could add to enamide to give the amido radical. The resulting amido radical could be easily oxidized to furnish the N-acyliminium. Upon deprotonation of N-acyliminium and tautomerization, the amidovinyl sulfone could be formed as the final product (Scheme 26).
C bonds to generate a-chlorosulfones (Scheme 26). Direct C–H functionalizations of enamides or enecarbamates under the optimized conditions proceeded with a wide scope of substrates and remarkable selectivity to give functionalized vinyl sulfones with good to excellent yields. They envisaged that sulfonyl radical, which could be generated from sulfonyl chloride under photocatalysis conditions, could add to enamide to give the amido radical. The resulting amido radical could be easily oxidized to furnish the N-acyliminium. Upon deprotonation of N-acyliminium and tautomerization, the amidovinyl sulfone could be formed as the final product (Scheme 26).
 |
| | Scheme 26 Visible-light photoredox-catalyzed sulfonation of alkenes. | |
3.4 Sulfonylation of alkenes with DMSO
In 2015, Yuan and Li48 provided a novel method via ammonium iodide-induced sulfonylation of alkenes to afford vinyl sulfone scaffolds utilizing DMSO as the versatile sulfur source (Scheme 27). Aromatic and heteroaromatic olefins proceeded smoothly to afford the target products. Also, interminal alkene and conjugated diene were productive, while allylbenzene failed to give the corresponding product. And the reaction was believed to mechanistically proceed via radical pathway.
 |
| | Scheme 27 Ammonium iodide-induced sulfonylation of alkenes. | |
3.5 Sulfonylation of alkenes with SO2
Recently, Willis developed a new convenient reagent DABSO (DABCO·SO2, the combination of DABCO and sulfur dioxide), which can serve as a surrogate of SO2 for the in situ formation of sulfinate salts in the synthesis of sulfonamides and sulfamides.49 In 2015, Feng and co-workers50 reported an efficient one-pot, three-component synthesis of vinyl sulfones via iodide-catalyzed radical alkenylation using aryl diazonium salts, terminal alkenes and DABSO (Scheme 28). The control experiments suggested a possible radical mechanism. First, TBHP decomposes to generate the tert-butoxyl and hydroxyl radicals with the assistance of the iodide anion. Secondly, the decomposition of the diazonium salts in the presence of t-BuO− forms the phenyl radical. Subsequently the addition of DABSO affords sulfonyl radical. Then, addition of sulfonyl radical to I2 formed sulfonyl halides. Sulfonyl halides easily generate corresponding sulfonyl radicals and cause atom transfer radical additions to multiple bonds. Alternatively, addition of sulfonyl radical to alkenes forms carbon-centered radical. The radical reacts with in situ generated iodine to generate β-iodosulfone. Finally, elimination of HI similar to the β-hydride elimination forms products to finish the catalytic cycle (Scheme 28).
 |
| | Scheme 28 Iodide-catalyzed radical alkenylation using aryl diazonium salts, terminal alkenes and DABSO. DABCO (1,4-diazabicyclo[2.2.2]octane), TABI (tetrabutylammonium iodide), DABSO (DABCO·SO2). | |
4. Decarboxylative sulfonylation of α,β-unsaturated carboxylic acids
4.1 Decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with sodium sulfinates
In the past decades, decarboxylative coupling reactions have been a prominent research topic because they provided straightforward and efficient pathways to the formation of carbon–carbon and carbon–heteroatom bonds under relatively mild conditions.51 Recently, a few approaches were described for converting α,β-unsaturated carboxylic acids to vinyl sulfones. In 2014, Pd-catalyzed decarboxylative cross-coupling reaction of cinnamic acid with arene sulfinic acid sodium salts for preparing vinyl sulfone scaffolds was reported by Tan and co-workers52 (Scheme 29). The reaction was found to tolerate both electron withdrawing and electron donating groups, affording various vinyl sulfones. Two possible reaction mechanisms for this coupling were also proposed (Scheme 29).
 |
| | Scheme 29 Pd-catalyzed decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with sodium sulfinates. dppb (1,4-bis(diphenylphosphino)butane). | |
In 2014, Guo and co-workers53 reported a Cu(II)-catalyzed decarboxylative sulfonylation of alkenyl carboxylic acids with sodium sulfinates using air as the oxidant (Scheme 30). It is the first aerobic decarboxylative sulfonylation of alkenyl carboxylic acids utilizing sodium sulfinates as the sulfur source without any silver additives for stereoselective synthesis of (E)-alkenyl sulfones. The mechanistic study indicates that the initial sulfonyl cation addition and the following decarboxylation processes are involved in this transformation (Scheme 30).
 |
| | Scheme 30 Cu-catalyzed decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with sodium sulfinates. | |
Subsequently, Prabhu and co-workers54 presented a efficient method for the preparing vinyl sulfones via CuII-catalyzed and ligand-promoted decarboxylative coupling of sodium aryl sulfinates with α,β-unsaturated acids (Scheme 31a). This decarboxylative radical coupling reaction proceeded exclusively to afford the (E)-isomer using a catalytic amount of Cu(ClO4)2·6H2O, 1,10-phenanthroline as a ligand, and TBHP in decane as an oxidant. Interestingly, using DMSO as the oxidant, a tandem cross-decarboxylative/coupling reaction for the preparation of vinyl sulfone scaffolds from cinnamic acid and sodium sulfinates under transition-metal-free conditions was reported by Jiang's group55 (Scheme 31b). The mechanism study of the reaction indicated that a radical pathway could be involved.
 |
| | Scheme 31 Decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with sodium sulfinates. | |
Very recently, Mao and Shi56 reported an iodine-promoted decarboxylative C–S bond formation through cinnamic acids and sodium benzene sulfinates in the absence of metal catalyst (Scheme 32a). In 2015, an efficient and green protocol for the preparation of (E)-vinyl sulfones via iodine-promoted decarboxylative cross-coupling reactions of sodium sulfinates with cinnamic acids using water as the solvent was developed by Yuan and co-workers57 (Scheme 32b). Kuhakarn'group58 also demonstrated the synthesis of (E)-vinyl sulfones via PhI(OAc)2 mediated decarboxylative sulfonylation strategy (Scheme 32c). A wide range of functionalities both in the aromatic and heteroaromatic unsaturated carboxylic acids and the sodium aryl sulfinates are tolerated with these reaction conditions. And the preliminary study on these reaction mechanisms also implies that these reactions are probably proceeding through a radical pathway.
 |
| | Scheme 32 I2 or PhI(OAc)2-promoted decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with sodium sulfinates. | |
Recently, Wang and Zha59 developed an electrochemical decarboxylative sulfono functionalization protocol for preparing (E)-vinyl sulfones directly from cinnamic acids and sodium sulfinates with high regioselectivity at room temperature (Scheme 33). In light of the experiments, a possible sulphonyl radical-based pathway was also discussed by the authors.
 |
| | Scheme 33 Electrochemical decarboxylative coupling between cinnamic acids and sodium sulfinates. | |
In 2015, Mao and Zhang60 reported a novel phosphoric acid-mediated synthesis of vinyl sulfones through decarboxylative coupling reactions of sodium sulfinates with phenylpropiolic acids (Scheme 34). A broad range of substrates, including both substituted propiolic acids and sodium sulfinates, were tolerated with this protocol under optimal conditions, and a variety of vinyl sulfone products were obtained in moderate to excellent yields. The plausible mechanism was also proposed (Scheme 34). An oxygen-centered radical was initially generated through the oxidation of sodium sulfinate by DMSO upon heating, which could be resonance-stabilized with a sulfonyl radical. Next, carbon-centered intermediate was formed through the trans addition of sulfonyl radical to the triple bond resulting from the deprotonation of phenylpropiolic acid in the presence of basic sodium sulfinate. An anionic was readily produced after the removal of one molecule of carbon dioxide followed by the abstraction of a hydrogen radical from phosphoric acid. With the assistance of phosphoric acid, the desired product was obtained.
 |
| | Scheme 34 Decarboxylative coupling between phenylpropiolic acid and sodium sulfinates. | |
4.2 Decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with sulfonylhydrazides
Arylsulfonyl hydrazides have evolved as excellent synthons in recent years and behave as a source for a sulfur nucleophile or electrophile depending upon the nature of the reaction conditions. Very recently, Singh and co-workers61 reported a novel metal-free protocol for room temperature decarboxylative sulfono functionalization using the reaction of sulfonyl hydrazides with cinnamic acids, to provide a range of vinyl sulfone molecules (Scheme 35). The reaction is presumed to involve a sulfonyl radical, which is formed by the sequential N–H abstraction by an iodine radical generated by the reaction of I2 with TBHP. The resulting sulfonyl radical subsequently undergoes addition to cinnamate followed by iodine catalyzed decarboxylation to afford (E)-vinyl sulfones (Scheme 35).
 |
| | Scheme 35 I2-catalyzed decarboxylative coupling between cinnamic acid and sulfonyl hydrazides. DBU, 1,8-diazabicyclo[5,4,0]undec-7-ene. | |
Interestingly, Mao and Zhang22 established a new approach to the selective synthesis of (E)-vinyl sulfones via a Fe/Cu co-catalyzed sulfonylation of arylpropiolic acids with sulfonyl hydrazides (Scheme 36a). Subsequently, Yang and Wu23 developed direct decarboxylative hydrosulfonylation of arylpropiolic acids with sulfonylhydrazides via copper-catalyzed under base-free conditions to obtain (E)-vinyl sulfones (Scheme 36b). This reaction features a simple catalytic system and good tolerance of substrates including aliphatic sulfonylhydrazides. Both the mechanism of the above two reactions are presumed to involve a sulfonyl radical, which is generated by Fe/Cu and DTBP or CuI and O2. And arylpropiolic acids can be transformed to alkynylcopper(I) by releasing CO2. The resulting sulfonyl radical subsequently undergoes addition to alkynylcopper(I) to afford (E)-vinyl sulfones.
 |
| | Scheme 36 Decarboxylative coupling between arylpropiolic acid and sulfonyl hydrazides. | |
4.3 Decarboxylative sulfonylation of α,β-unsaturated carboxylic acids with arylsulfinic acids
Very recently, encouraged by Mao and Zhang' work,22 Kuhakarn and co-workers62 reported Na2CO3-promoted decarboxylative sulfonylation of arylpropiolic acids with arylsulfinic acids in the absence of a transition metal catalyst (Scheme 37). This simple and environmentally benign transformation offers an alternative approach and allows for easy and rapid synthesis of (E)-vinyl sulfones. The preliminary study on the reaction mechanisms implies that the reaction is probably proceeding through a radical pathway.
 |
| | Scheme 37 Decarboxylative coupling between arylpropiolic acid and arylsulfonic acid. | |
5. Sulfonylation of vinyl halides, terminal epoxides, oxime acetates and β-nitrostyrenes
In recent years, more efficient methods using transition-metal catalysts, such as Pd or Cu, for the cross-coupling of sulfinate salts with allenyl boronic acids, vinyl bromides, or vinyl triflates for preparing vinyl sulfones were reported.63 But there are still filled with great challenges for the use of toxic metals or organic solvents. In 2013, a transition-metal-free route to (E)-vinyl sulfones from the coupling of sodium sulfinates with vinyl halides in water was reported by Chen and Yu64 (Scheme 38). The tentative mechanism might undergo through nucleophile addition of sodium sulfinate to phenylvinyl bromide and elimination of the bromine anion giving the desired product (Scheme 38).
 |
| | Scheme 38 Transition-metal-free coupling of vinyl halides with sodium sulfinates. | |
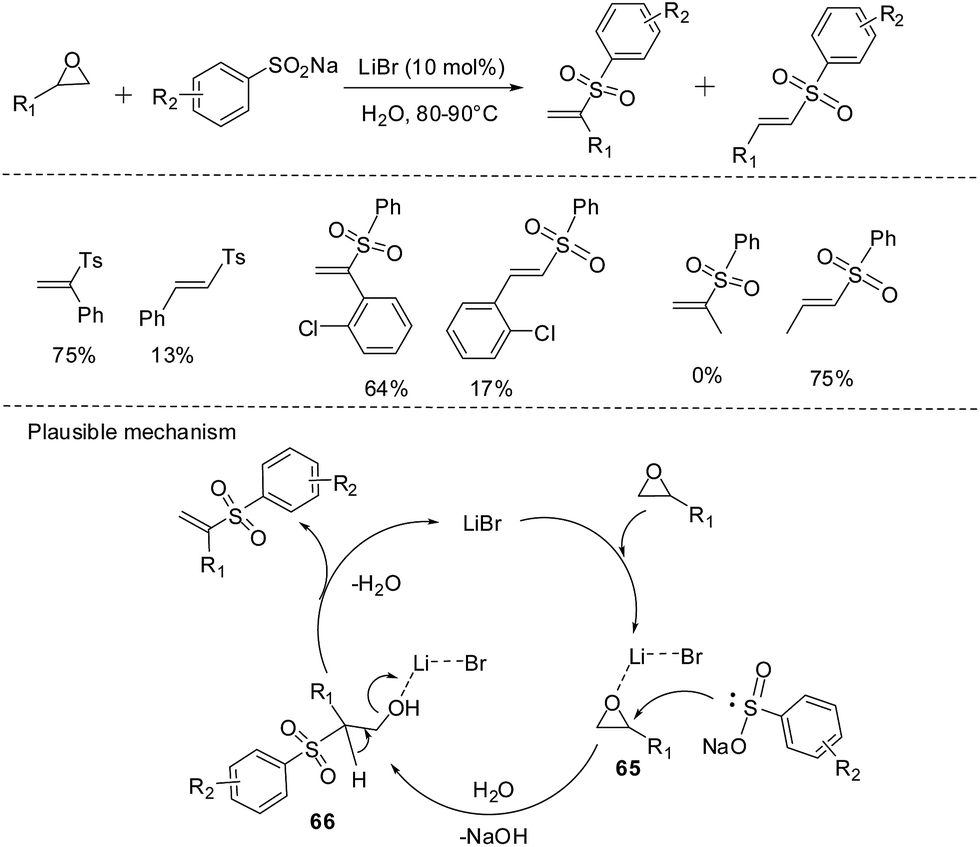
Epoxides, three-membered heterocycles, which can be used as starting materials as well as intermediates in order to carry out various synthetic transformations, possess some advantages, such as ease of preparation, ready accessibility in enantioriched forms65 and high reactivity towards a large variety of reagents, in particular nucleophiles.66 Recently, using water as the reaction solvent, an efficient regioselective preparation of vinyl sulfones catalysed by lithium bromide from sodium sulfinates and terminal epoxides in a one-pot procedure was disclosed by Yadav and co-workers.67 And this reaction gave two regioisomers (linear and branched vinyl sulfones) in most cases (Scheme 39). The plausible mechanism might undergoes through regioselective nucleophilic epoxide opening with the sulfinate group of sodium sulfinates and dehydrative β-elimination of β-hydroxy sulfones affording vinyl sulfones and liberating lithium bromide to complete the catalytic cycle.
 |
| | Scheme 39 Synthesis of vinyl sulfones from terminal epoxides and sodium sulfinates. | |
In 2014, a procedure involving CuII-catalyzed N–O bond cleavage, activation of a vinyl sp2 C–H bond, and C–S bond formation for synthesis of β-sulfonylvinylamines from sodium sulfinates and oxime acetates was reported by Jiang and co-workers68 (Scheme 40). One proposed mechanism is might undergo the CuII/CuI catalytic cycle through a single electron transfer process. Another possible mechanism might involve a key organocopperIII intermediate, which was proposed to occur via a CuII/CuIII catalytic cycle.
 |
| | Scheme 40 Synthesis of vinyl sulfones from oxime acetates and sodium sulfinates. | |
Very recently, an efficient silver-catalyzed one-pot protocol for the highly stereoselective synthesis of (E)-vinyl sulfones by denitrative radical cross-coupling of readily available β-nitrostyrenes and sodium sulfinates at room temperature under mild reaction conditions was investigated by Yadav and co-workers69 (Scheme 41). This is the first report on the facile formation of C(sp2)–S bonds leading to vinyl sulfones. And the reaction involves a radical addition/elimination pathway for the formation of the product.
 |
| | Scheme 41 Synthesis of vinyl sulfones from β-nitrostyrenes and sodium sulfinates. | |
6. Decomposition of tosylhydrazones
In recent years, N-tosylhydrazones, derived from simple ketones, which were utilized as the precursors of diazo compounds, have broad applications in constructing complex molecules by transition-metal-catalyzed or metal-free cross-coupling reactions.70 In 2014, Prabhu and co-workers71 revealed a novel reaction of tosylhydrazone with CNBr and aliphatic quaternary ammonium salts for preparing vinyl sulfones and witnessed the migration of a tosyl group for the first time (Scheme 42). They postulated that the reaction of tosylhydrazone with CNBr–TBAB to form vinyl sulfones proceeds through radical mechanism (Scheme 42). The tosyl group migration in these reactions has been further confirmed by performing a mixed reaction of two sulfonyl hydrazine derivatives with CNBr–TBAB under the optimal conditions, which yielded a mixture of vinyl sulfone products.
 |
| | Scheme 42 Synthesis of vinyl sulfones from tosylhydrazone. | |
Very recently, wang and co-workers72 described the first copper(II)-catalyzed radical reaction to synthesize various vinyl sulfones from readily available N-tosylhydrazones (Scheme 43). The transformation features excellent E stereoselectivity, broad substrate scope, low cost of reagents, and convenient operation. The plausible mechanism for the reaction is proposed (Scheme 43). The N-tosylhydrazone isomerized. Copper coordinated with C–C double bond and promoted the decomposition of N-tosylhydrazone releasing N2H2 and tosyl free radical to provide vinyl copper complex. Then, the tosyl free radical combined with vinyl copper complex to give copper carbenoid followed by the O–H insertion reaction with water to regenerate the copper catalyst and trans-elimination of H2O to afford vinyl sulfone.
 |
| | Scheme 43 Copper-catalyzed synthesis of vinyl sulfones from tosylhydrazone. | |
7. Conclusions and perspectives
During the last five years significant developments have been made on the synthesis of vinyl sulfones. Numerous new transition-metal or metal-free catalytic systems have been developed for improving the cross-coupling reactions of alkene or alkyne derivatives with sulfonyl derivatives. Many sulfonylating reagents such as sulfinic acids, sodium sulfinates and sulfonyl hydrazides, etc., have been explored for the direct sulfonylation of olefins and alkynes. In addition, the decarboxylative sulfonylation protocols of α,β-unsaturated carboxylic acids serve as an important complementary route to vinyl sulfones. Furthermore, the metal-free or transition-metal-catalyzed decomposition of tosylhydrazones provides a new avenue for synthesis of vinyl sulfone scaffolds. Mechanistic study could help us to understand the nature of these reactions. However, sulfonylation reaction of substrates containing an aromatic ring can proceed smoothly in most of these methodologies, whereas the corresponding reaction of heteroaromatic and aliphatic substrates suffer from some issues like poor activity or regioselectivity. Thus developing new methods to extend the substrate scope is highly demanding in this area. In addition, the exploration of robust catalytic systems will continue to drive this field. As a consequence, this review gives ample and updated information on the synthesis of this class of compounds and will be helpful in the development of improved methods for the synthesis of vinyl sulfones as well as other organosulfur compounds.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (21102003), the Natural Science Foundation of Anhui Province (1608085MB38), and the Program for the Introduction of Talent of Anhui University of Science & Technology.
References
-
(a) A. Vigalok, C-X Bond Formation, Springer, Heidelberg, 2010 CrossRef;
(b) A. K. Yudin, Catalyzed Carbon-Heteroatom Bond Formation, Wiley-VCH, Weinheim, 2010 CrossRef;
(c) H. Liu and X. Jiang, Chem.–Asian J., 2013, 8, 2546 CrossRef CAS PubMed.
-
(a) B. A. Frankel, M. Bentley, R. G. Kruger and D. G. McCafferty, J. Am. Chem. Soc., 2004, 126, 3404 CrossRef CAS PubMed;
(b) M. Uttamchandani, K. Liu, R. C. Panicker and S. Q. Yao, Chem. Commun., 2007, 1518 RSC;
(c) D. C. Meadows, T. Sanchez, N. Neamati, T. W. North and J. Gervay-Hague, Bioorg. Med. Chem., 2007, 15, 1127 CrossRef CAS PubMed;
(d) A. S. Newton, P. M. C. Glória, L. M. Gonçalves, D. J. V. A. dos Santos, R. Moreira, R. C. Guedes and M. M. M. Santos, Eur. J. Med. Chem., 2010, 45, 3858 CrossRef CAS PubMed;
(e) E. Dunny, W. Doherty, P. Evans, J. P. G. Malthouse, D. Nolan and A. J. S. Knox, J. Med. Chem., 2013, 56, 6638 CrossRef CAS PubMed;
(f) S. Y. Woo, J. H. Kim, M. K. Moon, S. H. Han, S. K. Yeon, J. W. Choi, B. K. Jang, H. J. Song, Y. G. Kang, J. W. Kim, J. Lee, D. J. Kim, O. Hwang and K. D. Park, J. Med. Chem., 2014, 57, 1473 CrossRef CAS PubMed;
(g) W. Doherty, J. James, P. Evans, L. Martin, N. Adler, D. Nolanb and A. Knox, Org. Biomol. Chem., 2014, 12, 7561 RSC.
-
(a) H. Kumamoto, K. Deguchi, T. Wagata, Y. Furuya, Y. Odanaka, Y. Kitade and H. Tanaka, Tetrahedron, 2009, 65, 8007 CrossRef CAS;
(b) Q. Zhu and Y. Lu, Org. Lett., 2009, 11, 1721 CrossRef CAS PubMed;
(c) O. Arjona, R. Menchaca and J. Plumet, Org. Lett., 2001, 3, 107 CrossRef CAS PubMed;
(d) M. N. Noshi, A. Elawa, E. Torres and P. L. Fuchs, J. Am. Chem. Soc., 2007, 129, 11242 CrossRef CAS PubMed;
(e) D. J. Wardrop and J. Fritz, Org. Lett., 2006, 8, 3659 CrossRef CAS PubMed;
(f) S. Sulzer-Mossé, A. Alexakis, J. Mareda, G. Bollot, G. Bernardinelli and Y. Filinchuk, Chem.–Eur. J., 2009, 15, 3204 CrossRef PubMed;
(g) A. López-Pérez, R. Robles-Machín, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2007, 46, 9261 CrossRef PubMed.
-
(a) M. Balasubramanian and V. Baliah, J. Chem. Soc., 1954, 1844 RSC;
(b) S. Chodroff and W. F. Whitmore, J. Am. Chem. Soc., 1950, 72, 1073 CrossRef CAS;
(c) D. A. R. Happer and B. E. Steenson, Synthesis, 1980, 10, 806 CrossRef.
-
(a) I. C. Popoff and J. L. Denver, J. Org. Chem., 1969, 34, 1128 CrossRef CAS;
(b) G. Wang, U. Mahesh, G. Y. J. Chen and S. Q. Yao, Org. Lett., 2003, 5, 737 CrossRef CAS PubMed;
(c) P. R. Sacasa, J. Zayas and S. F. Wnuk, Tetrahedron Lett., 2009, 50, 5424 CrossRef CAS PubMed;
(d) D. Dana, A. R. Davalos, G. Subramaniam, N. Afzal, W. H. Hersh and S. Kumar, Tetrahedron Lett., 2013, 54, 2717 CrossRef CAS.
-
(a) R. A. Gancarz and J. L. Kice, Tetrahedron Lett., 1980, 21, 4155 CrossRef CAS;
(b) M. Asscher and D. Vofsi, J. Chem. Soc., 1964, 4962 RSC;
(c) M. Asscher and D. Vofsi, J. Chem. Soc., Perkin Trans. 1, 1972, 1543 Search PubMed;
(d) P. B. Hopkins and P. L. Fuchs, J. Org. Chem., 1978, 43, 1208 CrossRef CAS.
-
(a) M. Kirihara, J. Yamamoto, T. Noguchi and Y. Hirai, Tetrahedron Lett., 2009, 50, 1180 CrossRef CAS;
(b) X. Huang, D. H. Duan and W. X. Zheng, J. Org. Chem., 2003, 68, 1958 CrossRef CAS PubMed.
-
(a) P. L. Fuchs and T. F. Braish, Chem. Rev., 1986, 86, 903 CrossRef CAS;
(b) I. Forristal, J. Sulfur Chem., 2005, 26, 163 CrossRef CAS;
(c) D. C. Meadows and J. Gervay-Hague, Med. Res. Rev., 2006, 26, 793 CrossRef CAS PubMed;
(d) J. C. Carretero and R. Gomez Arrayas, in Houben-Weyl Methods of Molecular Transformations. Science of Synthesis, ed. G. Molander, Georg Thieme Verlag, Stuttgart, 2007, vol. 23, p. 19 Search PubMed;
(e) T. Pathak, Tetrahedron, 2008, 64, 3605 CrossRef CAS;
(f) A. El-Awa, M. N. Noshi, X. M. du Jourdin and P. L. Fuchs, Chem. Rev., 2009, 109, 2315 CrossRef CAS PubMed.
-
(a) D. C. Reeves, S. Rodriguez, H. Lee, N. Haddad, D. Krishnamurthy and C. H. Senanayake, Tetrahedron Lett., 2009, 50, 2870 CrossRef CAS;
(b) H. Jiang, X. Chen, Y. Zhang and S. Yu, Adv. Synth. Catal., 2013, 355, 809 CrossRef CAS.
- Y. Xu, J. Zhao, X. Tang, W. Wu and H. Jiang, Adv. Synth. Catal., 2014, 356, 2029 CrossRef CAS.
- T. Sawangphon, P. Katrun, K. Chaisiwamongkhol, M. Pohmakotr, V. Reutrakul, T. Jaipetch, D. Soorukram and C. Kuhakarn, Synth. Commun., 2013, 43, 1692 CrossRef CAS.
- N. Taniguchi, Tetrahedron, 2014, 70, 1984 CrossRef CAS.
- A. Rodríguez and W. J. Moran, J. Org. Chem., 2016, 81, 2543 CrossRef PubMed.
- D. C. Craig, G. L. Edwards and C. A. Muldoon, Tetrahedron, 1997, 53, 6171 CrossRef CAS.
-
(a) D. H. R. Barton, M. S. Csiba and J. C. Jaszberenyi, Tetrahedron Lett., 1994, 35, 2869 CrossRef CAS;
(b) M. Yoshimatsu, M. Hayashi, G. Tanabe and O. Muraoka, Tetrahedron Lett., 1996, 37, 4161 CrossRef CAS.
- J. M. Fang and M. Y. Chen, Tetrahedron Lett., 1987, 28, 2853 CrossRef CAS.
- N. Mantrand and P. Renaud, Tetrahedron, 2008, 64, 11860 CrossRef CAS.
- S. F. Wang, C. P. Chuang, J. H. Lee and S. T. Liu, Tetrahedron, 1999, 55, 2273 CrossRef CAS.
- T. Taniguchi, A. Idota and H. Ishibashi, Org. Biomol. Chem., 2011, 9, 3135 Search PubMed.
- X. Li, X. Shi, M. Fang and X. Xu, J. Org. Chem., 2013, 78, 9499 CrossRef CAS PubMed.
- X. Li, X. Xu and X. Shi, Tetrahedron Lett., 2013, 54, 3071 CrossRef CAS.
- G. Rong, J. Mao, H. Yan, Y. Zheng and G. Zhang, J. Org. Chem., 2015, 80, 4697 CrossRef CAS PubMed.
- S. Li, X. Li, F. Yang and Y. Wu, Org. Chem. Front., 2015, 2, 1076 RSC.
-
(a) Q. Lu, J. Zhang, F. Wei, Y. Qi, H. Wang, Z. Liu and A. W. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156 CrossRef CAS PubMed;
(b) Q. Lu, J. Zhang, G. Zhao, Y. Qi, H. Wang and A. W. Lei, J. Am. Chem. Soc., 2013, 135, 11481 CrossRef CAS PubMed.
-
(a) J. M. Baskin and Z. Wang, Org. Lett., 2002, 4, 4423 CrossRef CAS PubMed;
(b) V. Nair, A. Augustine, T. G. George and L. G. Nair, Tetrahedron Lett., 2001, 42, 6763 CrossRef CAS;
(c) S. Cacchi, G. Fabrizi, A. Goggiamani, L. M. Parisi and R. Bernini, J. Org. Chem., 2004, 69, 5608 CrossRef CAS PubMed;
(d) D. C. Reeves, S. Rodriguez, H. Lee, N. Haddad, D. Krishnamurthy and C. H. Senanayake, Tetrahedron Lett., 2009, 50, 2870 CrossRef CAS;
(e) Q. L. Xu, L. X. Dai and S. L. You, Org. Lett., 2010, 12, 800 CrossRef CAS PubMed;
(f) N. Taniguchi, Synlett, 2011, 1308 CrossRef CAS;
(g) N. Taniguchi, Synlett, 2012, 1245 CrossRef CAS.
-
(a) R. Chawla, R. Kapoor, A. K. Singh and L. D. S. Yadav, Green Chem., 2012, 14, 1308 RSC;
(b) J. W. Lee, C. W. Lee, J. H. Jung and D. Y. Oh, Synth. Commun., 2000, 30, 2897 CrossRef CAS.
- Y. Xi, B. Dong, E. J. McClain, Q. Wang, T. L. Gregg, N. G. Akhmedov, J. L. Petersen and X. Shi, Angew. Chem., Int. Ed., 2014, 53, 4657 CrossRef CAS PubMed.
- W. Wei, J. Li, D. Yang, J. Wen, Y. Jiao, J. You and H. Wang, Org. Biomol. Chem., 2014, 12, 1861 Search PubMed.
- W. Wei, J. Wen, D. Yang, H. Jing, J. You and H. Wang, RSC Adv., 2015, 5, 4416 RSC.
- For selected recent examples of DMSO as reactant:
(a) X. Jiang, C. Wang, Y. Wei, D. Xue, Z. Liu and J. Xiao, Chem.–Eur. J., 2014, 20, 58 CrossRef CAS PubMed;
(b) G. Yuan, J. Zheng, X. Gao, X. Li, L. Huang, H. Chen and H. Jiang, Chem. Commun., 2012, 48, 7513 RSC;
(c) F. Luo, C. Pan, L. Li, F. Chen and J. Cheng, Chem. Commun., 2011, 47, 5304 RSC;
(d) L. Chu, X. Yue and F. L. Qing, Org. Lett., 2010, 12, 1644 CrossRef CAS PubMed;
(e) G. Yin, B. Zhou, X. Meng, A. Wu and Y. Pan, Org. Lett., 2006, 8, 2245 CrossRef CAS PubMed.
- Y. J. Jiang and T. P. Loh, Chem. Sci., 2014, 5, 4939 RSC.
- J. Y. Chen, X. L. Chen, X. Li, L. B. Qu, Q. Zhang, L. K. Duan, Y. Y. Xia, X. Chen, K. Sun, Z. D. Liu and Y. F. Zhao, Eur. J. Org. Chem., 2015, 314 CrossRef.
- G. Deng and J. Zou, ARKIVOC, 2010, ii, 186 Search PubMed.
- R. J. Song, Y. Liu, Y. Y. Liu and J. H. Li, J. Org. Chem., 2011, 76, 1001 CrossRef CAS PubMed.
- Q. Xue, Z. Mao, Y. Shi, H. Mao, Y. Cheng and C. Zhu, Tetrahedron Lett., 2012, 53, 1851 CrossRef CAS.
- Z. Liu, X. Chen, J. Chen, L. Qu, Y. Xia, H. Wu, H. Ma, S. Zhua and Y. Zhao, RSC Adv., 2015, 5, 71215 RSC.
-
(a) V. Varvoglis, Hypervalent Iodine in Organic Synthesis, Academic Press, London, 1997 Search PubMed;
(b) T. Wirth, Topics in Current Chemistry Vol. 224: Hypervalent Iodine Chemistry – Modern Developments in Organic Synthesis, Springer, Berlin, 2003 CrossRef.
-
(a) P. J. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS PubMed;
(b) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2002, 102, 2523 CrossRef CAS PubMed;
(c) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed.
- P. Katrun, S. Chiampanichayakul, K. Korworapan, M. Pohmakotr, V. Reutrakul, T. Jaipetch and C. Kuhakarn, Eur. J. Org. Chem., 2010, 5633 CrossRef CAS.
- B. Das, M. Lingaiah, K. Damodar and N. Bhunia, Synthesis, 2011, 18, 2941 CrossRef.
- N. Zhang, D. Yang, W. Wei, L. Yuan, Y. Cao and H. Wang, RSC Adv., 2015, 5, 37013 RSC.
- Y. C. Luo, X. J. Pan and G. Q. Yuan, Tetrahedron, 2015, 71, 2119 CrossRef CAS.
- A. U. Meyer, S. Jäger, D. P. Hari and B. König, Adv. Synth. Catal., 2015, 357, 2050 CrossRef CAS.
- F. C. Küpper, M. C. Feiters, B. Olofsson, T. Kaiho, S. Yanagida, M. B. Zimmermann, L. J. Carpenter, G. W. Luther, Z. Lu, M. Jonsson and L. Kloo, Angew. Chem., Int. Ed., 2011, 50, 11598 CrossRef PubMed.
- S. Tang, Y. Wu, W. Liao, R. Bai, C. Liu and A. Lei, Chem. Commun., 2014, 50, 4496 RSC.
- X. Li, Y. Xu, W. Wu, C. Jiang, C. Qi and H. Jiang, Chem.–Eur. J., 2014, 20, 7911 CrossRef CAS PubMed.
- H. Jiang, X. Chen, Y. Zhang and S. Yu, Adv. Synth. Catal., 2013, 355, 809 CrossRef CAS.
- X. Gao, X. Pan, J. Gao, H. Huang, G. Yuan and Y. Li, Chem. Commun., 2015, 51, 210 RSC.
- H. Woolven, C. Gonzalez-Rodriguez, L. Marco, A. L. Thompson and M. C. Willis, Org. Lett., 2011, 13, 4876 CrossRef CAS PubMed.
- W. Fan, J. Su, D. Shi and B. Feng, Tetrahedron, 2015, 71, 6740 CrossRef CAS.
-
(a) S. B. Lang, K. M. O'Nele and J. A. Tunge, J. Am. Chem. Soc., 2014, 136, 13606 CrossRef CAS PubMed;
(b) P. Fang, M. Li and H. Ge, J. Am. Chem. Soc., 2010, 132, 11898 CrossRef CAS PubMed;
(c) C. L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S. F. Zheng, B. J. Li and Z. J. Shi, Nat. Chem., 2010, 2, 1044 CrossRef CAS PubMed.
- R. Guo, Q. Gui, D. Wang and Z. Tan, Catal. Lett., 2014, 144, 1377 CrossRef CAS.
- Q. Jiang, B. Xu, J. Jia, A. Zhao, Y. R. Zhao, Y. Y. Li, N. N. He and C. C. Guo, J. Org. Chem., 2014, 79, 7372 CrossRef CAS PubMed.
- B. V. Rokade and K. R. Prabhu, J. Org. Chem., 2014, 79, 8110 CrossRef CAS PubMed.
- Y. Xu, X. Tang, W. Hu, W. Wu and H. Jiang, Green Chem., 2014, 16, 3720 RSC.
- J. Chen, J. Mao, Y. Zheng, D. Liu, G. Rong, H. Yan, C. Zhang and D. Shi, Tetrahedron, 2015, 71, 5059 CrossRef CAS.
- J. Gao, J. Lai and G. Yuan, RSC Adv., 2015, 5, 66723 RSC.
- P. Katrun, S. Hlekhlai, J. Meesin, M. Pohmakotr, V. Reutrakul, T. Jaipetch, D. Soorukram and C. Kuhakarn, Org. Biomol. Chem., 2015, 13, 4785 Search PubMed.
- P. Qian, M. Bi, J. Su, Z. Zha and Z. Wang, J. Org. Chem., 2016, 81, 4876 CrossRef CAS PubMed.
- G. Rong, J. Mao, H. Yan, Y. Zheng and G. Zhang, J. Org. Chem., 2015, 80, 7652 CrossRef CAS PubMed.
- R. Singh, B. K. Allam, N. Singh, K. Kumari, S. K. Singh and K. N. Singh, Org. Lett., 2015, 17, 2656 CrossRef CAS PubMed.
- J. Meesin, P. Katrun, V. Reutrakul, M. Pohmakotr, D. Soorukram and C. Kuhakarn, Tetrahedron, 2016, 72, 1440 CrossRef CAS.
-
(a) A. Battace, T. Zair, H. Doucet and M. Santelli, Synthesis, 2006, 3495 CAS;
(b) S. Cacchi, G. Fabrizi, A. Goggiamani, L. M. Parisi and R. Bernini, J. Org. Chem., 2004, 69, 5608 CrossRef CAS PubMed;
(c) M. Bian, F. Xu and C. Ma, Synthesis, 2007, 2951 CAS;
(d) F. Huang and R. A. Batey, Tetrahedron, 2007, 63, 7667 CrossRef CAS.
- S. Liang, R. Y. Zhang, G. Wang, S. Y. Chen and X. Q. Yu, Eur. J. Org. Chem., 2013, 7050 CrossRef CAS.
- R. M. Hanson, Chem. Rev., 1991, 91, 437 CrossRef CAS.
- A. S. Rao, S. K. Paknikar and J. G. Kirtane, Tetrahedron, 1983, 39, 2323 CrossRef CAS.
- R. Chawla, R. Kapoor, A. K. Singh and L. D. S. Yadav, Green Chem., 2012, 14, 1308 RSC.
- X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205 CrossRef CAS PubMed.
- T. Keshari, R. Kapoorr and L. D. S. Yadav, Eur. J. Org. Chem., 2016, 2695 CrossRef CAS.
-
(a) Z. Shao and H. Zhang, Chem. Soc. Rev., 2012, 41, 560 RSC;
(b) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed.
- D. P. Ojha and K. R. Prabhu, Org. Lett., 2015, 17, 18 CrossRef CAS PubMed.
- S. Mao, Y. R. Gao, X. Q. Zhu, D. D. Guo and Y. Q. Wang, Org. Lett., 2015, 17, 1692 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.