
Polyacrylates networks synthesized by endlinking of 3-armed precursor via radical addition coupling reaction†
Junmin Gao and
Qi Wang*
Key Laboratory of Macromolecular Synthesis and Functionalization, (Ministry of Education), Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310027, P. R. China. E-mail: wangq@zju.edu.cn
First published on 22nd June 2016
Abstract
We propose a strategy of synthesis of well-defined polyacrylate networks with cleavable branch units by endlinking of 3-armed prepolymer via radical addition coupling reaction with alkene. Via decrosslinking, cleavage at the branch units, the polymer network can be transformed to linear chains, which can be analyzed by normal methods. The length between the branch units, the distribution of the branch units and the fraction of the dangling chains of the network together with the extent of reaction are directly analyzed, which give detailed description of the polymer network. The method can be applied to prepare amphiphilic conetwork containing tert-butyl acrylate and acrylic acid units.
Introduction
Crosslinked polymers or polymer networks are an important class of polymers. The chemical and topological structure of the network determines its various properties. Radical copolymerization of vinyl monomer/multivinyl cross-linkers or random post-crosslinking of polymers with unsaturated double bonds produces polymer networks without control on crosslink density, distribution of branch units and ratio of dangling chains. Endlinking of end-functionalized linear or star polymers with multifunctional small molecules or with other end-functionalized star polymers is a promising method for synthesis of well-defined polymer networks.1,2 Since the precursor polymers are normally prepared by anionic polymerization, the resulting network chains exhibit a known average length and narrow molecular weight distribution. The crosslink functionality can be also adjusted by the functionality of the precursor polymer or the crosslinker. After occurrence of “click” chemistry,3 a number of networks have been synthesized by endlinking of end-functionalized star polymers based on copper-catalyzed azide–alkyne cycloaddition,4–8 catalyst-free azide–alkyne cycloaddition,9,10 thiol-ene coupling11–16 and Diels–Alder cycloaddition.17–20Although various “model” networks are claimed to be synthesized by the endlinking methodology, the polymer networks have not been proved to be modal by direct measurement. The main drawback of the endlinking method is that the average functionality of the branch points and the content of the dangling chain of the network remain unknown due to its insolubility, which supposedly is same as designed by the feed ratio. Even the “click” reactions of small molecules or linear polymer is believed to complete stoichiometrically, the corresponding reactions of multifunctional precursors resulting in nonlinear network is far from full conversion due to either the steric limitation or the gelation effect.
Polyacrylate networks can be prepared by radical copolymerization of acrylate and small amount of alkyl diacrylate as crosslinker. Although the crosslink density can be adjusted and indirectly measured, the distribution of the branch point remains unknown. “Modal” networks of poly(alkyl methacrylate) have been reported by endlinking of α,ω-bifunctional living precursor polymer prepared by anionic polymerization with ethylene dimethacrylate as crosslinker.21–24 For example, model networks were prepared by copolymerization of α,ω-living poly(glycidyl methacrylate) and α,ω-living poly(glycidyl methacrylate)-b-poly(methyl methacrylate)-b-poly(glycidyl methacrylate) triblock copolymers with ethylene dimethacrylate.23 No polyacrylate network was reported to be synthesized by endlinking of α,ω-living polyacrylate, but poly(acrylic acid) networks were prepared from α,ω-amino-terminated poly(acrylic acid) by cross-linking them with a trifunctional isocyanate coupling agent.25 Branched acrylic copolymers based on 2-hydroxypropyl acrylate were synthesized using ethylene glycol diacrylate, bisphenol A ethoxylated diacrylate or a disulfide-based diacrylate as branching comonomers by RAFT polymerization, and chemical degradation of the last branched acrylic copolymers produced thiol-functionalized primary chains.26 Modal network of poly(tert-butyl acrylate) (tBA) was prepared by endlinking of α,ω-azido-poly(tBA) with tri- and tetraacetylene cross-linkers by click reaction.5
The design and synthesis of “characterizable” polymer network is essential to understand the microstructure of polymer network. To the best of our knowledge, there is no report about full characterization of polymer network including crosslinking density, distribution of branching units and the content of dangling chain by direct experimental methods. As shown in Scheme 1, if the crosslinked polymer contains cleavable branch units, the polymer network can be transformed to linear chains after cleavage at branch units, which is defined as decrosslinkable polymer network. After decrosslinking, the resulting linear polymers can be characterized by normal methods and the structure of the network can be reconstructed. The length between the branch unit, the distribution of the branch unit and the content of the dangling chain of the network can be directly measured, which give the full description of the polymer network structure.
 | ||
| Scheme 1 Decrosslinkable PMA prepared by endlinking of 3-armed PMA and with α-methyl styrene via radical addition-coupling reaction. | ||
We previously reported the synthesis of AB-type diblock copolymers by coupling of two kinds of polymer precursors prepared by ATRP via radical addition cross-coupling reaction in presence of various double bonds.27,28 In this article, we propose the synthesis of well-defined decrosslinkable polyacrylate network by endlinking of 3-armed polyacrylate precursor bearing cleaved core with α-methyl styrene via radical addition-coupling reaction (RAC) as shown in Scheme 1 and the characterization of the polymer network.
Experimental section
The 3-armed poly(methyl acrylate) (tPMA) and poly(t-butyl acrylate) (tPtBA) were prepared by normal ATRP method using phenolic ester and alcohol ester trifunctional initiators respectively. tPMA or tPtBA, α-methyl styrene, PMDETA, Cu were reacted in THF under nitrogen. The product was firstly extracted with ethanol and THF until the gel was colorless. The sol part was obtained by removal of copper complex from the extracts by passing through a neutral alumina column. Both sol and gel parts were finally dried under vacuum at 40 °C. The gel and sol were treated with NaOH/methanol in THF for a certain time, yielding cleaved sol and gel. Full descriptions of the methods, as well as GPC and NMR data were given in the ESI.†Results and discussion
(1) Radical addition-coupling reaction between monobromo poly(methyl acrylate) (PMA-Br) and different alkenes
Here, efficient endlinking of PMA-Br via radical coupling reaction is impossible, because polyacrylate-type radical prefers disproportionation rather than coupling.29 The ATRC of PMA-Br promoted by Cu(0)/2,2′-bipyridine30 and Cu(0)/CuBr with PMDETA, Me6TREN and 1,4,8,11-tetraazacyclotetradecane31 were reported and the coupling efficiency varied from 14% to 54%. We studied the RAC of linear monobromo PMA-Br with different alkenes as model reaction. PMA-Br prepared by ATRP (Mn,GPC = 1240 g mol−1, PDI = 1.20) and 0.5 equivalent of α-methyl styrene (AMS), 1,1-diphenyl ethylene (DPE) or styrene were reacted with Cu(0)/PMDETA at 50 °C for 4 h. The RAC reaction of PMA-Br includes two steps shown in Scheme 2. The first step is the addition of PMA radical (PMA˙) in situ generated by single-electron-transfer (SET) to carbon–carbon double bond of alkene, which generates intermediate radical (PMA-alkene˙). The second step is the cross-coupling of PMA˙ and PMA-alkene˙, which produces diblock polymer (PMA-alkene–PMA). The obtained product was measured by GPC and fitted with two Gaussian functions (see Fig. S1–S4†). One peak is derived from diblock polymer formed by RAC, the other is mono-block polymer formed by disproportionation, chain-transfer reaction or other termination reactions as shown in Scheme 2. The coupling efficiency can be calculated based on the peak area ratio, which is equal to Sdiblock/Stotal. According to the results listed in Table 1, the coupling efficiency for three alkenes are between 54% and 67%. Among three alkenes, AMS gives the highest coupling efficiency under the same condition. In the presence of more than 1 equivalent of styrene, the coupling efficiency of PMA-Br under ATRC condition can be as high as 99% due to the self-coupling of styrene-terminated radical (PMA-St˙),31 which is different from cross-coupling in this paper (PMA-St˙ + PMA˙). Coupling efficiency of 60% was obtained in the presence of 0.5 equiv. of styrene,31 which is very close to our result. | ||
| Scheme 2 Radical addition-coupling reaction (RAC) between linear PMA-Br and alkene. | ||
(2) Polymer network synthesized by radical addition-coupling polymerization using 3-armed PMA and α-methyl styrene
The 3-armed PMA (tPMA) was prepared by ATRP using a trifunctional initiator 1,3,5-(2′-bromo-2′-methylpropionato)benzene.32 Phenolic ester-based initiator was used to facilitate the transesterification of the core of PMA network without changing the side ester groups. Under the condition [tPMA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[AMS]:[Cu]:[PMDETA] = 1:1.5:3.6:3.6, [tPMA] = 0.02 M, 50 °C, the branched and crosslinked polymers were prepared depending on the polymerization time or the extent of reaction (p).
:[AMS]:[Cu]:[PMDETA] = 1:1.5:3.6:3.6, [tPMA] = 0.02 M, 50 °C, the branched and crosslinked polymers were prepared depending on the polymerization time or the extent of reaction (p).
The product after gelation was firstly extracted with ethanol and THF until the gel was colorless. Then, the sol part was obtained by removal of copper complex from the extracts by passing through a neutral alumina column. Both sol and gel parts were finally dried under vacuum at 40 °C.
The optimal condition for cleavage reaction of branch unit was obtained by using tPMA as the sample. The tPMA (Mn,GPC = 4070 g mol−1, PDI = 1.09, Mn,NMR = 4830 g mol−1) was treated with NaOH/THF/methanol at 60 °C for 2 hours. The resulting linear PMA was measured by GPC and the number average molecular weight (Mn) and polydispersity index are 1210 g mol−1 and 1.26 (see Fig. S5†). By comparison of the 1H-NMR spectra of tPMA and its cleaved product shown in Fig. 1, it was found that aromatic proton corresponding to phenolic ester group totally disappeared in the spectrum of cleaved product and the alcoholic ester groups remained unchanged. The average molecular weight of cleaved product calculated by 1H-NMR was Mn,NMR = 1560 g mol−1, very close to one third of the Mn,NMR of tPMA. This suggests the transesterification only occurs at the ester group of the initiator. Such condition was applied to the cleavage reaction of both sol and gel fraction and the they were transformed into linear PMA after transesterification reaction.
 | ||
| Fig. 1 1H-NMR spectrum of 3-armed PMA (bottom) and its cleaved product (top). | ||
The sol, cleaved sol and gel were characterized by GPC. As shown in Fig. 2, the GPC curve of the cleaved gel present two peaks, which correspond to the dangling chains and network chains of crosslinked PMA derived from uncoupled and coupled arms of tPMA respectively. The molecular weight of the network chain (Mn,net = 4180 g mol−1, PDInet = 1.07) was almost the twice of the dangling chain (Mn,dangling = 1960 g mol−1, PDIdangling = 1.07) according to resolving the GPC curve with two Gaussian functions. The weight fraction of dangling chain of gel part (Fd,w) can be calculated based on the two peak area, which is Fd,w = Sdangling/Stotal. Mn,net corresponds to the crosslink density and PDInet describes the distribution of the branch unit (Db). The extent of reaction for gel part (pgel) can be estimated by peak area ratio of network chains to total chains (pgel = Snet/Stotal). The sol fraction can be treated and analyzed by the same method. If the weight fractions of sol (wsol) and gel (wgel) are available, the extent of reaction of the whole system (ptotal) is given by (psol × wsol + pgel × wgel).
 | ||
| Fig. 2 GPC curves of cleaved gel part of crosslinked PMA (run 3 in Table 1) and fitting results. | ||
The extent of reaction, p, is defined as the conversion of the functional group in stepwise polymerization and can be obtained by end-group analysis. Our current method estimates the value of p by analysis of resultant linear polymer. The fitting peak with high molecular weight only corresponds to network chains (PMA–AMS–PMA), while fitting peak with low molecular weight includes contribution from PMA with bromo-, saturated or unsaturated ends and PMA–AMS due to only slight difference in molecular weight. The p evaluated by our method is slightly lower than true value, because the PMA–AMS is treated as unreacted arm same as PMA.
The related data of sol and gel parts of crosslinked PMA obtained at different times are summarized in Table 2. The gelation occurred at around 80 minutes and the pc = 0.666. After gelation, the reaction continued and the maximum extent of reaction pmax reached 0.718.
| Run | Time (min) | Sol fraction | Gel fraction | Total | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mwb | PDIb | psolc | wsold | pgelc | wgeld | Fd,we | ptotalf | ||
| a Polymerization condition: [tPMA]:[AMS]:[Cu]:[PMDETA] = 1:1.5:3.6:3.6, [tPMA] = 0.02 M, 50 °C.b Number- and weight-average molecular weight (Mn, Mw) in 103 g mol−1 and polydispersity index (PDI) of sol measured by GPC.c The extent of reaction measured by peak fitting of the GPC curves of cleaved products, p = Snet/Stotal.d The weight faction of sol and gel parts.e The weight faction of dangling chain.f p = psol × wsol + pgel × wgel. |
|||||||||
| 1 | 40 | 50.7 | 3.25 | 0.465 | 1 | — | — | — | 0.465 |
| 2 | 50 | 57.0 | 3.52 | 0.545 | 1 | — | — | — | 0.545 |
| 3 | 81 | 20.0 | 2.0 | 0.371 | 0.2 | 0.742 | 0.80 | 0.258 | 0.666 |
| 4 | 96 | 14.4 | 1.67 | 0.489 | 0.14 | 0.756 | 0.86 | 0.244 | 0.718 |
The coupling efficiency of 3-armed polymer precursor is higher than that of linear one, although the reaction conditions are same for two kinds of precursors. In the linear RAC, the viscosity of the reaction medium does not change so much since the molecular weight of the product is not so high. In the non-linear RAC, the viscosity increases greatly around gel point. The high viscosity normally hinders the diffusion of the macromolecules and significantly slows the reaction between two macro-radicals, such as disproportionation of PMA macro-radicals, but hardly affects the addition reaction between macro-radical PMA˙ and AMS. Compared with macro-radical PMA˙, the intermediate radical PMA–AMS˙ generated by addition reaction shown in Scheme 2 is much stable. Hence, this leads to higher coupling efficiency in the non-linear RAC than the linear RAC. The same explanation has been applied to the autoacceleration in radical addition polymerization at high conversion.29
(3) Amphiphilic polymer conetwork containing tert-butyl acrylate and acrylic acid units
Following the same protocol, crosslinked poly(tert-butyl acrylate) (PtBA) was prepared. The method involves the endlinking of 3-armed PtBA-Br (Mn,GPC = 6900 g mol−1, PDI = 1.06, Mn,NMR = 4770 g mol−1) prepared by ATRP using a trifunctional initiator (1,1,1-tris(2-bromoisobutyryloxymethyl)propane) (TBBMP). The alcohol ester-based initiator instead of phenolic ester was used to facilitate the hydrolysis of tBA unit without change of ester groups of branch units derived from the initiator. The gel part was partially hydrolyzed by trifluoroacetic acid at room temperature for 24 hours, which generated crosslinked copolymer containing tBA and acrylic acid (AA) units (Scheme 3). The conetwork presents amphiphilic property and the swelling ratios in three different solvents are given in Table 3. The network can be fully hydrolyzed and cleaved by NaOH in water affording linear poly(acrylic acid) (Scheme 3), which is evidenced by disappearance of tert-butyl groups in its 1H-NMR spectra (see Fig. S13†).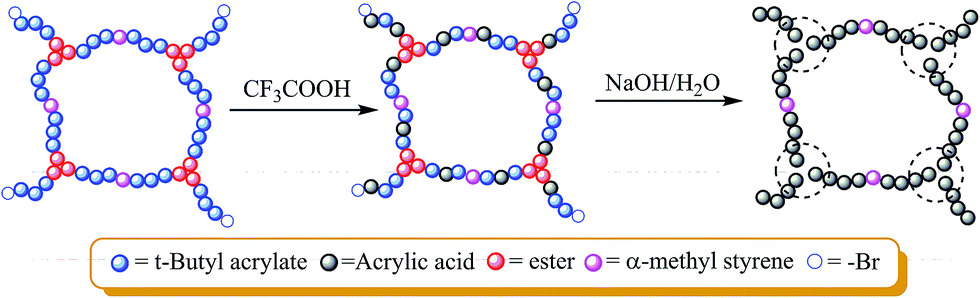 | ||
| Scheme 3 Partial hydrolysis, total hydrolysis and decrosslinking of P(t-butyl acrylate) gel. | ||
Model network of poly(acrylic acid) have been prepared by endlinking of α,ω-amino-terminated poly(acrylic acid) with tris(4-isocyanatophenyl)methane.25 Our method based on RAC of macroradical generated by bromo-polymer provides a simple preparative method of polymer networks. Multi-armed polymers can be easily prepared by ATRP33 and used directly without end-group transform, which is required in endlinking of two kinds of polymer precursors by “click” reactions.
Conclusions
We present synthesis of polyacrylate networks by endlinking of 3-armed polyacrylate prepolymers via RAC reaction in the presence of alkene. The obtained networks are decrosslinkable and experimental accessible, which allows the full characterization of the complicated structure of the network. The crosslink density (Mn,net), the distribution of the branch unit (Db) and the fraction of the dangling chain (Fd) can be obtained directly by GPC measurement of cleaved product. The protocol can be applied to prepare amphiphilic crosslinked copolymer containing tert-butyl acrylate and acrylic acid units.Acknowledgements
Financial support from National Natural Science Foundation of China (21274120) is appreciated.Notes and references
- G. Hild, Prog. Polym. Sci., 1998, 23, 1019–1149 CrossRef CAS.
- P. Rempp and J. E. Herz, Angew. Makromol. Chem., 1979, 76, 373–391 CrossRef.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 26, 2774–2776 RSC.
- J. A. Johnson, D. R. Lewis, D. Az, M. G. Finn, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 6564–6565 CrossRef CAS PubMed.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromolecules, 2007, 40, 3589–3598 CrossRef CAS.
- M. H. Samiullah, D. Reichert, T. Zinkevich and J. Kressler, Macromolecules, 2013, 46, 6922–6930 CrossRef CAS.
- H. W. Ooi, K. S. Jack, H. Peng and A. K. Whittaker, Polym. Chem., 2013, 4, 4788–4800 RSC.
- J. A. Johnson, J. M. Baskin, C. R. Bertozzi, J. T. Koberstein and N. J. Turro, Chem. Commun., 2008, 26, 3064–3066 RSC.
- C. A. DeForest, E. A. Sims and K. S. Anseth, Chem. Mater., 2010, 22, 4783–4790 CrossRef CAS PubMed.
- M. P. Lutolf and J. A. Hubbell, Biomacromolecules, 2003, 4, 713–722 CrossRef CAS PubMed.
- N. Gupta, B. F. Lin, L. Campos, M. D. Dimitriou, S. T. Hikita, N. D. Treat, M. V. Tirrell, D. O. Clegg, E. J. Kramer and C. J. Hawker, Nat. Chem., 2010, 2, 138–145 CrossRef CAS PubMed.
- J. Cui, M. A. Lackey, A. E. Madkour, E. M. Saffer, D. M. Griffin, S. R. Bhatia, A. J. Crosby and G. N. Tew, Biomacromolecules, 2012, 13, 584–588 CrossRef CAS PubMed.
- S. Grube and W. Oppermann, Macromolecules, 2013, 46, 1948–1955 CrossRef CAS.
- K. Oberg, Y. Hed, I. J. Rahmn, J. Kelly, P. Lowenhielm and M. Malkoch, Chem. Commun., 2013, 49, 6938–6940 RSC.
- H. W. Ooi, K. S. Jack, A. K. Whittaker and H. Peng, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4626–4636 CAS.
- H. Zhou, J. Woo, A. M. Cok, M. Wang, B. D. Olsen and J. A. Johnson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19119–19124 CrossRef CAS PubMed.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Biomacromolecules, 2013, 14, 949–953 CrossRef CAS PubMed.
- A. M. Cok, H. Zhou and J. A. Johnson, Macromol. Symp., 2013, 329, 108–112 CrossRef CAS.
- H. Zhou and J. A. Johnson, Angew. Chem., Int. Ed., 2013, 52, 2235–2238 CrossRef CAS PubMed.
- A. Haeringe, G. Hild, P. Rempp and H. Benoit, Macromol. Chem. Phys., 1973, 169, 249–260 CrossRef.
- R. Okasha and G. Hild, C. R. Acad. Sci., 1978, 287, 97–99 CAS.
- G. Hild and J. P. Lamps, Polymer, 1995, 36, 4841–4850 CrossRef CAS.
- G. Hild and P. Rempp, C. R. Acad. Sci., 1969, 269, 1622–1625 CAS.
- A. Shefer, A. J. Grodzinsky, K. L. Prime and J. P. Busnel, Macromolecules, 1993, 26, 5009–5014 CrossRef CAS.
- C. D. Vo, J. Rosselgong, S. P. Armes and N. C. Billingham, Macromolecules, 2007, 40, 7119–7125 CrossRef CAS.
- C. Y. Zhang and Q. Wang, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2817–2823 CrossRef CAS.
- Q. Q. Zhu and Q. Wang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2029–2036 CrossRef CAS.
- G. Odian, Principles of Polymerization, Wiley & Sons, Inc., Hoboken, New Jersey, 4th edn, 2004 Search PubMed.
- B. Otazaghine, C. Boyer, J. J. Robin and B. Boutevin, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2377–2394 CrossRef CAS.
- T. Sarbu, K. Y. Lin, J. Spanswick, R. R. Gil, D. J. Siegwart and K. Matyjaszewski, Macromolecules, 2004, 37, 9694–9700 CrossRef CAS.
- D. M. Haddleton and C. Waterson, Macromolecules, 1999, 32, 8732–8739 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GPC and the 1H-NMR spectra of obtained polymers. See DOI: 10.1039/c6ra10703f |
| This journal is © The Royal Society of Chemistry 2016 |