
Preparation of magnetic nanographene sorbent for extraction and quantification of targeted PPCPs in environmental water samples†
Masoumeh Rashvand,
Maryam Vosough* and
Kazem Kargosha
Chemistry and Chemical Engineering Research Center of Iran, P. O. Box 14335-186, Tehran, Iran. E-mail: vosough@ccerci.ac.ir; Fax: +98 21 44787707; Tel: +98 21 44787714
First published on 2nd August 2016
Abstract
In this study, a novel graphene-based magnetic nanocomposite sorbent was prepared via one-step co-precipitation method and its chemical structure and morphology was studied. This novel sorbent was employed for the first time for extraction of seven pharmaceutical and personal care products (PPCPs) in complex environmental water samples prior to high performance liquid chromatography with diode-array detection. Benefiting both nanographene sorbent and magnetic phase separation, provided some advantages such as fast separation, high adsorption capacity, low sorbent amount and solvent consumption in comparison with traditional methods. To achieve the optimum condition for the magnetic solid phase extraction (MSPE) procedure, various effective extraction parameters including mass of sorbent, desorption solvent volume and desorption time were optimized using central composite design (CCD). Under the optimal situation, the recoveries ranged from 70 up to 120% for majority of validation samples and relative standard deviations (RSDs) varied between 1.2% and 9.5%. The limits of detection for targeted analytes were in the range of 0.02 to 0.9 μg L−1. Finally, the proposed method was successfully applied to complex environmental samples with acceptable recoveries.
1. Introduction
During recent years, different methods have been developed and adopted for sample preparation prior to instrumental trace and ultra-trace analysis. Solid phase extraction (SPE) as a sorption-based sample preparation technique, is among the most widely used in analytical laboratories and is commonly used to clean-up and pre-concentrate the analytes.1–6 Besides SPE advantages, such as relatively low consumption of organic solvent, reproducibility, wide variety of the solid sorbents, high enrichment factors and good recoveries, it may suffer from a few drawbacks like time consuming procedure and in the case of dispersive mode, phase separation difficulties. To overcome such limitations, the employment of magnetic materials as the adsorbent has recently gained increasing research interest.7–10 The retrieval and separation process in magnetic solid phase extractions (MSPE) can be accomplished benefitting magnetically modified adsorbents and using an external magnet without any further centrifugation or filtration.Graphene, a two dimensional plannar sheet of carbon atoms, due to its unique properties, such as good mechanical, thermal and chemical stability, electron rich structure and large specific surface area, has been utilized as a versatile sorbent in solid-phase-based extraction techniques.11–16 The nano composite magnetic graphene oxide (MGO) could be prepared via various approaches such as the co-precipitation method,17–19 the solvothermal method,20–23 the hydrothermal method24–27 and the covalent bonding method.28,29
There are a few reports on the coupling of the magnetic properties with the characteristics of graphene oxide, and implementing the resulting sorbent, i.e. MGO, in an extraction procedure.30–34 Wang et al.35 investigated the potential of MGO for removal of fuchsine dye from aqueous solution. Removing trace levels of polybrominated diphenyl ethers (BDEs) were studied in water treatment by employing the Fe3O4@PDDA/GOx@DNA composite absorbent.36 Another research was done by Luo et al.37 on determination of organochlorine pesticide (OCP) residues in tobacco. MGO was used as an adsorbent in this work and final determination was performed by on-line gel permeation chromatography-gas chromatography-tandem mass spectroscopy. As another example for pesticide residue analysis. Li et al.38 used MGO for determination of carbamate pesticides in tomatoes. Parabens in cosmetic products were also determined using magnetic nano composite of self-doped polyaniline–graphene by Mehdinia et al.39 Moreover, nano magnetic graphene oxide is also realized for sorption of metal ions. Kazemi et al.40 applied MGO for separation and pre-concentration of ultra-trace amount of gold ions. Co(II), Ni(II), Cu(II), Cd(II) and Pb(II) have been also determined by Sun et al.41 in biological samples using magnetic SPE pretreatment procedure followed by ICP-MS. In the work by Bao et al.42 a thiol-functionalized magnetic/graphene oxide (MGO) hybrid exhibited high adsorption capacity for Hg(II) removal. The sorption of Eu(III) on MGO was studied by Li43 and the result indicated that MGO would be acceptable candidate for removal of Eu(III) from aquatic environmental samples. In another work by Li,44 the sequestration of Sr(II) and Cs(I) was investigated using MGO in aqueous solutions. Besides, the adsorption investigation, the interaction mechanism between Sr(II)/Cs(I) and magnetic graphene oxides was highlighted in this study.
As emerging contaminant and due to the ecotoxicological and human health risks associated with their occurrence in the environment, pharmaceutical and personal care products (PPCPs) have received growing attention.45–47 PPCPs include diverse categories of medicals and ingredients of daily personal care products, which are utilized in high amounts and based on the reports have been monitored and detected in aquatic environments throughout the world.48–50 Continuous exposure to PPCPs can create significant impacts, even at low concentration. Fent et al.51 reported chronic toxicity of UV filter and some narcotics and also, endocrine disrupting effects of parabens were studied by Dobbins and Yamomoto.52,53 Due to the conventional designs of the sewage treatment plants (STPs), in which selective or specific removal of the individual contaminants are not taken into account, considerable classes of pharmaceutical and personal care products have been detected in effluents.54–58 On the other hand, some of these contaminants are discharged directly into rivers without any treatment via domestic wastewater, especially in rural regions. Therefore, it is essential to study the occurrence of mentioned pollutants in the receiving media of the STPs.
In the present study, a selected set of PPCPs, i.e. ethyl paraben (Et-P), propyl paraben (Pro-P), butyl paraben (But-P), benzophenone 3 (BZ-3), 4-methylbenzilidine camphor (4-MBC), diclofenac (Dic) and ibuprofen (Ibu) were analysed in multiple effluents of STPs and receiving rivers. Parabens are typical preservatives which frequently appear in the formulation of PCPs, and BZ-3 and 4-MBC are UV-filters, used in sunscreen products. Diclofenac and ibuprofen are categorized as non-steroidal anti-inflammatory drugs and have prevalent applications.
In the present work, the potential application of nano magnetic graphene oxide due to its large specific surface area and reusability property was investigated for the enrichment of selected PPCPs, for the first time. Although, the common methods for synthesis of MGO are solvothermal synthesis and covalent bonding, because of their complexity and rigorous conditions, the co-precipitation procedure was chosen in this case. Optimization of extraction parameters was carried out using central composite design (CCD) and finally the analytes were quantified by HPLC with diode array detection.
2. Materials and methods
2.1. Reagents and materials
Analytical standard of diclofenac and ibuprofen were donated by Tehran Darou Pharmaceuticals (Iran). The paraben standards were obtained from Sigma-Aldrich (USA). Benzophenone-3, 4-methylbenzilidine camphor were purchased from DSM (Netherlands). Hydrochloric acid (37%), sulfuric acid (98%), ammonia solution and potassium chlorate were obtained from Merck (Germany). HPLC-grade methanol (MeOH) and acetonitrile (ACN) were from Loba Chemie (India) and the reagents FeCl3·6H2O, FeCl2·4H2O and acetic acid (98%) were from Fluka (Switzerland). All aqueous solutions were prepared with ultrapure water provided by a Milli-Q purification system from Millipore (USA). Solvents, calibration and real samples were filtered through 0.22 μm nylon filter membranes filter paper from Varian (USA).Stock solutions of selected PPCPs were prepared by dissolving a specific weight of each pure component in methanol and kept in amber vials in the freezer (−18 °C). Working standards were made daily in mobile phase composition by dilution of stock solutions and stored in the dark at 4 °C. Calibration set contained 15 samples involving Et-P, Pro-P, But-P (0.2–15 μg L−1), Ibu (3–30 μg L−1), Dic (0.5–15 μg L−1), BZ-3 and 4-MBC (0.5–25 μg L−1) and were prepared in 5 mL volumetric flasks.
2.2. Apparatus
The HPLC analyses were performed on an Agilent 1200 series system (Agilent Technologies Inc., USA), equipped with a Rheodyne 7725 manual injector with a 50 μL injection loop, a degasser system, a quaternary pump, a column oven compartment, a Hewlett-Packard 1200 series photo diode-array detector (DAD) and Chemstation software package (version B.03.01), for controlling the instrument, data acquisition and data handling. A C18 column (15 cm × 0.46 cm, 5 μm particle size) was employed for chromatographic separation.X-ray diffraction system (Bruker AXS-D8 Advanced, Germany) at the step size of 2° per second was used for characterized of Fe3O4/GO composite. The morphology of GO and magnetic GO was investigated employing field emission scanning electron microscope (Sigma VP, ZEISS, Germany) and atomic force microscopy (FemtoScan, Russia). Chemical structure of samples was checked using Fourier transform infrared spectroscopy (Perkin Elmer, USA) and energy dispersive X-ray spectroscopy (Sirius SD, England).
2.3. Synthesis of MGO
Graphene oxide was synthesized according to the literature:59 1 g of graphite was added to 27 mL of concentrated sulfuric acid/nitric acid mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume, respectively) while stirring in an ice bath. Then, 11 g of potassium chloride was added gradually to complete oxidation. Standing over 4 days, the GO product was separated by centrifugation and washed several times with Milli-Q water to remove the residual acid. Finally, the product was dried at 60 °C under vacuum.
:1 by volume, respectively) while stirring in an ice bath. Then, 11 g of potassium chloride was added gradually to complete oxidation. Standing over 4 days, the GO product was separated by centrifugation and washed several times with Milli-Q water to remove the residual acid. Finally, the product was dried at 60 °C under vacuum.
Fe3O4/GO was synthesized thorough chemical co-precipitation procedure of Fe(II) and Fe(III) at the molar ratio of 1:2.60 90 mg of synthesized GO was exfoliated in 50 mL of water via ultra-sonication for about 1 h, then a 50 mL of iron source solution containing FeCl3·6H2O (800 mg) and FeCl2·4H2O (300 mg) in DI water was added gradually with vigorous stirring. The solution was heated to 85 °C and ammonia solution was added to raise pH to 10 and to precipitate the Fe(II) and Fe(III) ions. Afterwards, the solution was stirred rapidly and then allowed to cool down to room temperature. Finally, the resulting precipitate was separated using external magnet and washed repeatedly with Milli-Q water and dried at 60 °C.
2.4. Sample collection
Effluent wastewater samples were collected from three sewage treatment plants (Mahallati, Ertebatat and Rah) and three hospitals (Kasra, Razi and Pastor no) in Tehran (Iran). River samples were taken from Kan, Karaj and Jajrud rivers. Tap and dam waters also collected from a suburb of Tehran as the real samples. Groundwater samples were collected from different regions of Tehran (Iran).2.5. Analytical procedure
All real samples were centrifuged (Universal 320R, 4000 rpm) and filtered through 0.22 μm nylon membrane filters. For MSPE process, 25 mg of MGO was transferred into a tube and dispersed in 30 mL sample. Then the tube was sonicated (Sonorex Digital 10P) for about 5 min and shaken (IKA Shaker, KS 20) for 10 min. Subsequently, the sorbent was separated using an external magnetic field and the supernatant was easily discarded. Afterwards, 3 mL of acetonitrile was added to the MGO nanoparticles and shaken for 5 min. By the application of external magnet, the supernatant was collected and transferred into a 4 mL conical vial and evaporated under the stream of nitrogen. Finally, after re-dissolving the residue in 500 μL of mobile phase and being passed through a 0.22 μm PTFE syringe filter, the solution was introduced to HPLC/DAD for quantification.A gradient elution program, using (A) water (containing 0.3% acid acetic), (B) acetonitrile and (C) methanol was implemented for separation by HPLC. Elution started at 30:30:40 composition of A:B:C with flow rate 1.2 mL min−1 and reached to 10:30:60 composition over 4 min. As can be seen in Fig. 1, all analytes were eluted in 4.2 min. DAD data was recorded between 210 and 400 nm with the spectral resolution of 2 nm and integration period of 0.4 s per spectrum.
 | ||
| Fig. 1 Chromatogram of a standard mixture of seven PPCPs at concentration of 15 μg L−1 for Ibu and 4 μg L−1 for the rest at wavelengths 225 nm (red dotted line) and 270 nm (black solid line). | ||
3. Result and discussion
3.1. Characterization of MGO
To ascertain the successful synthesis of the sorbent, the X-ray diffraction pattern of GO and MGO was investigated. According to Fig. 2A, a sharp peak at 14° represents the crystalline structure of GO. The main diffraction peaks at 2θ = 30.16, 35.68, 43.46, 54.6, 57.28 and 62.76 which corresponding to the characteristic crystalline spinel structure of Fe3O4, are observed in XRD pattern of MGO.61 | ||
| Fig. 2 (A) XRD pattern of GO and MGO, (B) AFM image of GO, FESEM images of (C) GO and (D) MGO. | ||
Furthermore, the morphology and structure of GO and MGO were studied by FESEM and AFM. The AFM image of GO sheets in Fig. 2B demonstrates the full exfoliation of graphite oxide by cross-section analysis (thickness ∼ 0.94 nm).62 Moreover, the layer-like structure of graphene oxide can be clearly seen in Fig. 2C. Also nano particles of Fe3O4 that decorated the surface of GO are indicated in Fig. 2D.
Regarding the chemical structure of the synthesized sorbent, FT-IR spectra of GO and MGO were recorded (Fig. S1†). In both spectra, the absorptions bands at 1631, 3435 cm−1 are attributed to stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and OH groups, respectively. In addition, the bands associated with C–O stretching of the epoxy and the C–O stretching of carboxyl groups could be seen at 1051 and 1401 cm−1. Although the main absorption bands of their spectra are similar, the adsorption band at 596 cm−1 which is attributed to stretching vibration of Fe–O indicates the existence of Fe3O4. Besides, the energy-dispersive X-ray spectroscopy (EDX) result confirms the composition of synthesized sorbent and demonstrated the existence of Fe, C and O elements. The first peak (C) is mainly due to the basal plane of graphene oxide, the O peak is attributed to both Fe3O4 and the oxygen-containing functional groups of graphene oxide. The typical Fe peaks in the range between 6 and 7 keV confirmed the presence of Fe-oxide nanoparticles on the GO surface (Fig. 3).
C and OH groups, respectively. In addition, the bands associated with C–O stretching of the epoxy and the C–O stretching of carboxyl groups could be seen at 1051 and 1401 cm−1. Although the main absorption bands of their spectra are similar, the adsorption band at 596 cm−1 which is attributed to stretching vibration of Fe–O indicates the existence of Fe3O4. Besides, the energy-dispersive X-ray spectroscopy (EDX) result confirms the composition of synthesized sorbent and demonstrated the existence of Fe, C and O elements. The first peak (C) is mainly due to the basal plane of graphene oxide, the O peak is attributed to both Fe3O4 and the oxygen-containing functional groups of graphene oxide. The typical Fe peaks in the range between 6 and 7 keV confirmed the presence of Fe-oxide nanoparticles on the GO surface (Fig. 3).
 | ||
| Fig. 3 EDX result of MGO. | ||
3.2. Multivariate optimization of the extraction parameters
The experimental design can be useful for optimization of the parameters and understanding the interaction between effective factors and their effects on extraction performance. However, before designing and performing the experiments, pH of the extraction media and its effect on the extraction efficiency was studied, separately.The reason was that eliminating one of the factors, would significantly reduce the number of the designed experiments. Also, pH was not expected to have any interaction with the other selected factors (Table 1). So, it was evaluated in a one-at-a-time manner and the pH = 5 was showed to be the optimum value for all of the targeted analytes. In the next step, a central composite design (CCD) was employed for optimization of the significant factors of MDSPE process.
| Factors | Unit | Experimental field | |
|---|---|---|---|
| Minimum value (−1) | Maximum value (+1) | ||
| Mass of sorbent | mg | 10 | 30 |
| Desorption time | min | 2 | 5 |
| Desorption volume | mL | 1 | 3 |
CCD contains an embedded factorial or fractional factorial design with center point that is augmented with an additional design, often a star design, that allows estimation of the probable curvature.63 In this regard, based on preliminary experiments and the literature, sorbent mass (A), desorption solvent volume (B) and desorption time (C) were considered as the main factors of the MDSPE procedure (Table 1). Also, the peak areas of individual analytes and their geometric mean in every run were selected as responses for model development and optimization procedure. Totally, 20 experiments were designed by CCD as could be found in details with corresponding responses in Table S1.†
Due to the analysis of variance (ANOVA) on the global desirability (i.e. the geometric mean of the individual analytes responses), the model was significant and the parameters A, C, AB, AC, A2, B2, C2 and ABC were significant. Statistical parameters, such as coefficient of variation (4.97%), adequate precision (12.67), R2 (0.97), RAdj2 (0.92), RPred2 (0.83), low values of prediction error sum of squares (PRESS) and non-significant lack-of-fit (p = 0.7485), revealed the reliability of the proposed model. The optimum values of significant factors at the maximum global desirability were defined as 25 mg for mass of sorbent, 3 mL for desorption solvent volume and 5 min for desorption time. Fig. 4 shows the 3D response surface plots by defining the global desirability as the response. The influence of sorbent mass and desorption solvent volume could be seen in Fig. 4a while the third variable was set at its optimal value. So, by increasing the sorbent mass, the desirability was increased, as long as more volume of desorption solvent was used. In a similar way, Fig. 4b and c depict the effect of sorbent mass and desorption time and also, the effect of desorption time and desorption solvent volume on desirability function, respectively. Based on these plots, raising the solvent volume and desorption time would increase the global desirability, due to complete desorption and elution. The desirability also increases with increasing sorbent mass to 25 mg. The negative effect of increasing sorbent mass above 25 mg could be as a result of aggregation of sorbent particles and decreasing the effective surface.
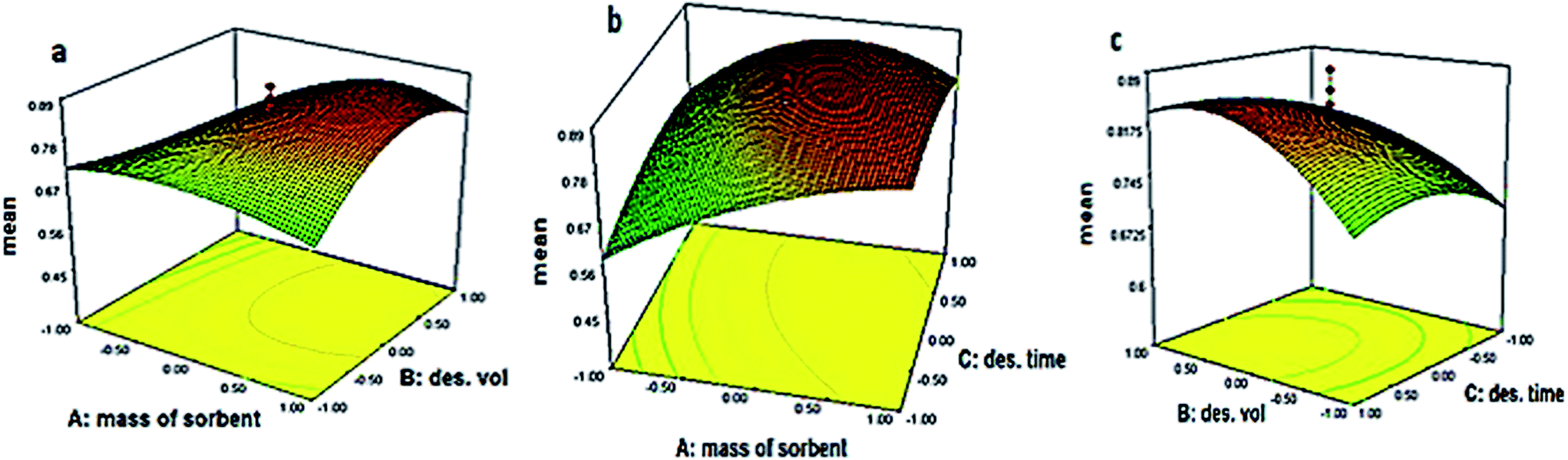 | ||
| Fig. 4 3D response surface plots of global desirability changing when optimizing (a) mass of sorbent–desorption volume, (b) mass of sorbent–desorption time and (c) desorption volume–desorption time while the third factor set at the optimum value. | ||
3.3. Validation of the method
Validation samples were made by spiking 5 representative real samples with the analytes at four concentration levels (Table S2†) and used to evaluate the accuracy and precision of the proposed method. So, the entire procedure of sample preparation and chromatographic analysis, under the optimized set of conditions, was implemented for all validation samples. The chromatogram of a spiked STP sample (STP-Eff, S3) can be seen in Fig. 5. Most of the analytes were quantified via univariate calibration by signal detection at 258 nm for parabens, 282 nm for Dic, 288 nm for BZ-3 and 300 nm for 4-MBC. A substantial baseline drift, due to the gradient mobile phase programming, affected quantification of Ibu at wavelength 225 nm. In fact, the recoveries of Ibu by signal detection at 225 nm were in the range of 10.9% to 35.6% for the two higher spike concentration levels. For the other samples, no clear peak was observed and quantification of Ibu was not successful using conventional univariate method. | ||
| Fig. 5 Chromatogram of a spiked sample (STP-Eff, S3) at wavelengths 225 nm (red dotted line) and 270 nm (black solid line). | ||
To overcome this problem, the data files were exported as ASCII format and then a background correction method based on the proposed strategy by Eilers,64 was employed, in which a background elimination algorithm for two-way signals was used, based on asymmetric least square spline regression. The effect of background correction on the chromatographic region containing Ibu peak can be appreciated from Fig. S2B which is the same plot of Fig. S2A† after correcting the background signal. As can be seen from this figure, a signal enhancement has been achieved by eliminating noise and background components. Finally, peak area calculation was performed as a multivariate scenario using well-known method of multivariate curve resolution-alternating least squares (MCR-ALS).65
The predicted concentration values together with analytical performance characteristics of the method have been shown in Table 2. As can be seen in this table, recovery of the extraction procedure for the validation set, with different matrix characteristics, for the majority of validation samples ranged between 70% and 120%. For the lowest spike level of Ibu and BZ-3 in real samples, the recoveries below 70% were obtained, that might be due to the noticeable baseline for Ibuprofen (Fig. S2A†) and low peak intensity for BZ-3.
| Spiked samples | Component (μg L−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| Et-P | Pro-P | But-P | Dic | Ibua | BZ-3 | 4-MBC | ||
| a The concentration values obtained using multivariate modelling. | ||||||||
| STP-Eff | S1 | 1.00(100.1%) | 0.84(84.0%) | 1.10(110.1%) | 2.49(124.5%) | 2.63(65.7%) | 1.16(58.0%) | 1.90(95.0%) |
| S2 | 1.64(82.0%) | 1.65(82.5%) | 1.67(83.5%) | 2.81(93.7%) | 7.07(88.4%) | 4.33(108.2%) | 2.71(90.3%) | |
| S3 | 4.39(109.7%) | 3.71(92.8%) | 4.42(110.5%) | 3.25(81.2%) | 11.89(79.3%) | 8.82(110.2%) | 2.93(73.2%) | |
| S4 | 9.52(95.2%) | 8.74(87.4%) | 9.32(93.2%) | 10.58(105.8%) | 20.77(83.1%) | 16.34(81.7%) | 9.12(91.2%) | |
| RSD (%) | 7.6 | 7.1 | 1.2 | 7.4 | 8.1 | 2.4 | 7.6 | |
| Hospital-Eff | S1 | 0.81(81.3%) | 0.80(80.1%) | 0.74(74.2%) | 2.42(121.2%) | 2.54(63.5%) | 1.39(69.5%) | 1.40(70.0%) |
| S2 | 1.61(80.5%) | 1.51(75.5%) | 1.61(80.5%) | 3.34(111.3%) | 6.94(86.8%) | 3.34(83.5%) | 20.20(73.3%) | |
| S3 | 3.17(79.3%) | 3.13(78.3%) | 3.17(79.3%) | 4.38(109.5%) | 13.69(91.3%) | 8.91(111.4%) | 3.10(77.5%) | |
| S4 | 8.35(83.5%) | 8.19(81.9%) | 8.55(85.5%) | 10.86(108.6%) | 23.42(93.7%) | 19.94(99.7%) | 8.09(80.9%) | |
| RSD (%) | 6.8 | 7.2 | 2.8 | 7.9 | 9.3 | 3.2 | 6.6 | |
| River | S1 | 0.85(85.5%) | 1.17(117.3%) | 0.98(97.6%) | 1.91(95.5%) | 2.73(68.2%) | 1.14(57.0%) | 1.34(67.0%) |
| S2 | 1.65(82.5%) | 1.73(86.5%) | 1.44(72.0%) | 2.59(86.3%) | 7.16(89.5%) | 4.77(119.2%) | 2.21(73.7%) | |
| S3 | 4.06(101.5%) | 3.68(92.0%) | 4.22(105.5%) | 2.99(74.7%) | 10.84(72.3%) | 8.71(108.9%) | 3.21(80.2%) | |
| S4 | 9.98(99.8%) | 10.3(103.1%) | 10.1(101.0%) | 9.13(91.3%) | 20.40(81.6%) | 23.90(119.5%) | 7.01(70.1%) | |
| RSD (%) | 6.5 | 6.9 | 1.7 | 7.1 | 8.3 | 2.1 | 6.9 | |
| Well | S1 | 0.95(94.9%) | 0.89(88.9%) | 1.02(101.9%) | 1.96(98.0%) | 2.77(69.2%) | 1.09(54.5%) | 1.50(75.0%) |
| S2 | 1.76(88.0%) | 1.73(86.5%) | 1.66(83.0%) | 2.44(81.3%) | 5.99(74.9%) | 4.09(102.3%) | 2.44(81.3%) | |
| S3 | 4.09(102.3%) | 3.73(93.2%) | 4.24(106.0%) | 3.05(76.3%) | 12.25(81.7%) | 7.66(95.8%) | 2.90(72.5%) | |
| S4 | 9.14(91.4%) | 9.71(97.1%) | 9.20(92.0%) | 7.93(79.3%) | 26.20(104.8%) | 20.90(104.5%) | 8.58(85.8%) | |
| RSD (%) | 6.1 | 6.4 | 1.4 | 6.9 | 9.5 | 2.8 | 7.2 | |
| Tap | S1 | 1.04(104.3%) | 1.25(125.4%) | 1.16(115.9%) | 1.38(69.0%) | 2.89(72.2%) | 1.31(65.5%) | 2.02(101.0%) |
| S2 | 2.07(103.4%) | 1.56(78.0%) | 1.57(78.5%) | 2.40(80.0%) | 5.81(72.6%) | 5.01(125.3%) | 3.22(107.4%) | |
| S3 | 4.47(111.8%) | 3.94(98.5%) | 4.54(113.5%) | 3.09(77.3%) | 14.74(98.3%) | 8.15(101.9%) | 3.27(81.7%) | |
| S4 | 7.76(77.6%) | 8.03(80.3%) | 8.43(84.3%) | 8.85(88.5%) | 23.42(93.7%) | 15.94(79.7%) | 8.67(86.7%) | |
| RSD (%) | 6.7 | 5.8 | 1.2 | 6.3 | 7.8 | 2.4 | 6.2 | |
| R2 | 0.996 | 0.997 | 0.993 | 0.998 | 0.991 | 0.993 | 0.990 | |
| LOD (μg L−1) | 0.03 | 0.02 | 0.03 | 0.13 | 0.9 | 0.18 | 0.07 | |
| LOQ (μg L−1) | 0.09 | 0.06 | 0.09 | 0.39 | 2.7 | 0.54 | 0.21 | |
| SEN | 8.92 | 12.30 | 8.45 | 2.47 | 1.22 | 2.23 | 7.93 | |
| Linear range (μg L−1) | 0.2–15 | 0.2–15 | 0.2–15 | 0.5–15 | 3–30 | 0.5–25 | 0.5–25 | |
Precision of the method was evaluated based on the repeatability of the analyses of selected PPCPs in spiked samples. The resulting relative standard deviations (RSDs) varied between 1.2% and 9.5% for triplicate extraction of water samples spiked at four concentration levels of S1 to S4 (Table 2), indicating a good level of repeatability. Furthermore, the limit of detection (LOD), defined as the concentrations at signal to noise ratio (S/N) of 3, limit of quantification (LOQ), on the basis of S/N ratio of 10 and sensitivity could be observed in Table 2. All the parameters illustrated the capability of the method for complex real samples.
3.4. Method comparison
The analytical performance of the proposed method was compared with the reported methods, based on different pretreatment strategies, for analysis of the selected PPCPs in similar matrixes. As demonstrated in Table 3, detection limits of the selected analytes in this procedure are lower than other methods. Also, eight consecutive adsorption/desorption cycles, no significant efficiency loss was observed. In addition, using magnetic sorbent facilitated the extraction procedure and shortened the MDSPE process time.| Determination technique | Analyte | Extraction technique | LOD (μg L−1) | Matrix | Ref. |
|---|---|---|---|---|---|
| a Bar adsorptive microextraction.b Solvent assisted dispersive.c Hollow fiber-liquid phase microextraction.d Ultra-high performance supercritical fluid chromatography.e Ultrasound-assisted ionic liquid dispersive liquid–liquid microextraction. | |||||
| HPLC-DAD | ET-P, Pro-P, But-P | BA-μEa | 0.1 | Water | 66 |
| GC-MS | ET-P, Pro-P, But-P | DLLME | 2.5–22 | Water | 67 |
| GC-PID | ET-P, Pro-P, But-P | SADb-SPME | 0.05–0.3 | River water | 68 |
| HPLC | BZ-3 | DLLME | 0.5 | Swimming pool water | 69 |
| HPLC-UV | BZ-3 | DLPME | 0.8 | Swimming pool water | 70 |
| HPLC-DAD | Dic-Ibu | HF-LPMEc | 52.9–40.6 | Urine | 71 |
| UHPSFCd | Dic-Ibu | US-IL-DLLMEe | 2.26–2.56 | Tap water | 72 |
| HPLC-DAD | ET-P, Pro-P, But-P | MDSPE | 0.02–0.03 | Water samples | This work |
| BZ-3–4-MBC | 0.18–0.07 | ||||
| Dic-Ibu | 0.13–0.9 | ||||
3.5. Real sample analysis
To demonstrate the applicability the proposed method for real samples, the selected contaminants were analysed in several environmental water samples, such as effluents of STPs and hospitals, river water and groundwater samples. Based on the results (Table 4), ethyl paraben, diclofenac and 4-MBC were found in most of the real samples.| Real samples | PPCPs (μg L−1) | |||||||
|---|---|---|---|---|---|---|---|---|
| Et-P | Pro-P | But-P | Dic | Ibu | BZ-3 | 4-MBC | ||
| STP-Eff | Mahallati | 0.43 | 0.09 | 0.19 | 0.51 | <LOQ | 1.49 | 1.39 |
| Ertebatat | 0.69 | <LOQ | <LOQ | 0.55 | <LOQ | <LOQ | 1.12 | |
| Rah | 0.19 | 0.20 | <LOQ | 0.49 | <LOQ | 0.89 | 1.28 | |
| Hospital-Eff | Kasra | 0.82 | 0.26 | 0.09 | 5.03 | <LOQ | <LOQ | 1.21 |
| Razi | 5.54 | 0.16 | 0.22 | 0.46 | <LOQ | <LOQ | 1.13 | |
| Pastor no | 0.18 | <LOQ | 0.21 | 0.92 | <LOQ | <LOQ | 1.22 | |
| Rivers | Karaj | 0.09 | 0.57 | <LOQ | 0.39 | <LOQ | <LOQ | 1.35 |
| Kan | 0.33 | 0.06 | 0.16 | 0.54 | <LOQ | 0.53 | 0.99 | |
| Jajrood | <LOQ | <LOQ | <LOQ | 0.46 | <LOQ | 0.72 | 1.32 | |
| Wells | Well1 | 0.16 | <LOQ | <LOQ | 0.52 | <LOQ | <LOQ | 1.07 |
| Well2 | 0.15 | <LOQ | <LOQ | 0.55 | <LOQ | <LOQ | 1.99 | |
| Well3 | 0.32 | 0.20 | 0.16 | <LOQ | <LOQ | <LOQ | 0.85 | |
| Tap | <LOQ | <LOQ | <LOQ | 0.42 | <LOQ | <LOQ | 0.24 | |
| Dam | 0.16 | <LOQ | <LOQ | 0.50 | <LOQ | 0.75 | 1.44 | |
On the contrast, propyl paraben, butyl paraben and BZ-3 were rarely observed in these samples. Besides, ibuprofen could not be quantified in any unspiked samples. This could be related to higher removal efficiency throughout the wastewater treatment processes and the lower stability of Ibu in aquatic samples. The results revealed an acceptable performance of the proposed method in terms of repeatability, limits of detection, sensitivity, accuracy and recovery for analysis of the selected PPCPs in real environmental samples.
4. Conclusion
In this paper, the applicability of magnetic graphene oxide was investigated for extraction of a selected group of PPCPs. The MGO sorbent was synthesized through a chemical co-precipitation procedure and its morphology and chemical structure was characterized using FESEM, XRD, FT-IR, EDX and AFM. The novel sorbent was implemented in a series of optimization experiments to study and find the best MDSPE conditions for the selected pollutants. The proposed method, regarding its acceptable figures of merit and recovery values, low solvent consumption and cost effectiveness, was satisfactory employed for quantification of mentioned analytes in environmental samples. Results indicated the existence of the majority of targeted pollutants in examined samples, confirming the importance of improving the efficiency of treatment processes and also, monitoring programs for PPCPs.References
- N. Fontanals, R. M. Marce and F. Borrull, J. Chromatogr. A, 2007, 1152, 14–31 CrossRef CAS PubMed.
- M. Farré, L. Kantiani, M. Petrovic, S. Pérez and D. Barceló, J. Chromatogr. A, 2012, 1259, 86–99 CrossRef PubMed.
- M. Padron, C. Afonso-Olivares, Z. Sosa-Ferrera and J. Santana-Rodriguez, Molecules, 2014, 19, 10320–10349 CrossRef PubMed.
- K. Wille, H. F. D. Brabander, L. Vanhaecke, E. De Wulf, P. V. Caeter and C. R. Janssen, TrAC, Trends Anal. Chem., 2012, 35, 87–108 CrossRef CAS.
- E. D. Guerrero, R. N. Marín, R. C. Mejías and C. G. Barroso, J. Chromatogr. A, 2007, 1167, 18–26 CrossRef CAS PubMed.
- L. Ramos, J. Chromatogr. A, 2012, 1221, 84–98 CrossRef CAS PubMed.
- R. A. Pérez, B. Albero, J. L. Tadeo, E. Molero and C. Sánchez-Brunete, Anal. Bioanal. Chem., 2015, 407, 1913–1924 CrossRef PubMed.
- L. Wang, X. Xu, Z. Zhang, D. Zhang, X. Liu and L. Zhang, RSC Adv., 2015, 5, 22022–22030 RSC.
- H. Abdolmohammad-Zadeh and Z. Talleb, Talanta, 2015, 134, 387–393 CrossRef CAS PubMed.
- A. Samadi and M. Amjadi, Microchim. Acta, 2015, 182, 257–264 CrossRef CAS.
- Y. B. Luo, G. T. Zhu, X. S. Li, B. F. Yuan and Y. Q. Feng, J. Chromatogr. A, 2013, 1299, 10–17 CrossRef CAS PubMed.
- Q. Liu, J. Shi and G. Jiang, TrAC, Trends Anal. Chem., 2012, 37, 1–11 CrossRef CAS.
- Y. B. Luo, Z. G. Shi, Q. Gao and Y. Q. Feng, J. Chromatogr. A, 2011, 1218, 1353–1358 CrossRef CAS PubMed.
- Y. B. Luo, B. F. Yuan, Q. W. Yu and Y. Q. Feng, J. Chromatogr. A, 2012, 1268, 9–15 CrossRef CAS PubMed.
- Y. B. Luo, J. S. Cheng, Q. Ma, Y. Q. Feng and J. H. Li, Anal. Methods, 2011, 3, 92–98 RSC.
- Y. Q. Feng and Y. B. Luo, Chin. J. Chromatogr., 2011, 29, 947–948 CAS.
- Y. Sun, W. Zhang, H. Yu, C. Hou, D. S. Li, Y. Zhang and Y. Liu, J. Alloys Compd., 2015, 638, 182–187 CrossRef CAS.
- C. Liang, T. Zhai, W. Wang, J. Chen, W. Zhao, X. Lu and Y. Tong, J. Mater. Chem. A, 2014, 2, 7214–7220 CAS.
- P. N. Diagboya, B. I. Olu-Owolabi and K. O. Adebowale, RSC Adv., 2015, 5, 2536–2542 RSC.
- C. Santhosh, P. Kollu, S. Doshi, M. Sharma, D. Bahadur, M. T. Vanchinathan, P. Saravanan, B. S. Kim and A. N. Grace, RSC Adv., 2014, 4, 28300–28308 RSC.
- Q. H. Wu, B. Qu, J. Tang, C. Wang, D. Wang, Y. Li and J. G. Ren, Electrochim. Acta, 2015, 156, 147–153 CrossRef CAS.
- A. Mehdinia, N. Khodaee and A. Jabbari, Anal. Chim. Acta, 2015, 868, 1–9 CrossRef CAS PubMed.
- L. Jing, A. Fu, H. Li, J. Liu, P. Guo, Y. Wang and X. S. Zhao, RSC Adv., 2014, 4, 59981–59989 RSC.
- Y. Dong, K. C. Yung, R. Ma, X. Yang, Y. S. Chui, J. M. Lee and J. A. Zapien, Carbon, 2015, 86, 310–317 CrossRef CAS.
- A. Hu, X. Chen, Q. Tang and B. Zeng, Ceram. Int., 2014, 40, 14713–14725 CrossRef CAS.
- S. Yang, L. Chen, L. Mu and P. C. Ma, J. Colloid Interface Sci., 2014, 430, 337–344 CrossRef CAS PubMed.
- F. Zhang, Y. Song, S. Song, R. Zhang and W. Hou, ACS Appl. Mater. Interfaces, 2015, 7, 7251–7263 CAS.
- P. S. Teo, H. N. Lim, N. M. Huang, C. H. Chia and I. Harrison, Ceram. Int., 2012, 38, 6411–6416 CrossRef CAS.
- A. Hu, X. Chen, Y. Tang, Q. Tang, L. Yang and S. Zhang, Electrochem. Commun., 2013, 28, 139–142 CrossRef CAS.
- Q. Wu, M. Liu, X. Ma, W. Wang, C. Wang, X. Zang and Z. Wang, Microchim. Acta, 2012, 177, 23–30 CrossRef CAS.
- P. Shi and N. Ye, Talanta, 2015, 143, 219–225 CrossRef CAS PubMed.
- A. Pourjavadi, Z. Tehrani and S. Jokar, J. Ind. Eng. Chem., 2015, 28, 45–53 CrossRef CAS.
- A. Speltini, M. Sturini, F. Maraschi, L. Consoli, A. Zeffiro and A. Profumo, J. Chromatogr. A, 2015, 1379, 9–15 CrossRef CAS PubMed.
- H. Teymourian, A. Salimi and S. Khezrian, Biosens. Bioelectron., 2013, 49, 1–8 CrossRef CAS PubMed.
- C. Wang, C. Feng, Y. Gao, X. Ma, Q. Wu and Z. Wang, Chem.–Eur. J., 2011, 173, 92–97 CAS.
- N. Gan, J. Zhang, S. Lin, N. Long, T. Li and Y. Cao, Materials, 2014, 7, 6028–6044 CrossRef CAS.
- Y. B. Luo, X. Li, X. Y. Jiang, B. D. Cai, F. P. Zhu, H. F. Zhang, Z. G. Chen, Y. Q. Pang and Y. Q. Feng, J. Chromatogr. A, 2015, 1406, 1–9 CrossRef CAS PubMed.
- N. Li, J. Chen and Y. Shi, Talanta, 2015, 141, 212–219 CrossRef CAS PubMed.
- A. Mehdinia, R. Esfandiarnejad and A. Jabbari, J. Sep. Sci., 2015, 38, 141–147 CrossRef CAS PubMed.
- E. Kazemi, S. Dadfarnia and A. M. Shabani, Talanta, 2015, 141, 273–278 CrossRef CAS PubMed.
- J. Sun, Q. Liang, Q. Han, X. Zhang and M. Ding, Talanta, 2015, 132, 557–563 CrossRef CAS PubMed.
- J. Bao, Y. Fu and Z. Bao, Nanoscale Res. Lett., 2013, 8, 486–491 CrossRef PubMed.
- D. Li, B. Zhang and F. Xuan, J. Mol. Liq., 2015, 211, 203–209 CrossRef CAS.
- D. Li, B. Zhang and F. Xuan, J. Mol. Liq., 2015, 209, 508–514 CrossRef CAS.
- J. M. Brausch and G. M. Rand, Chemosphere, 2011, 82, 1518–1532 CrossRef CAS PubMed.
- P. Gago-Ferrero, M. S. Díaz-Cruz and D. Barceló, Anal. Bioanal. Chem., 2012, 404, 2597–2610 CrossRef CAS PubMed.
- J. L. Liu and M. H. Wong, Environ. Int., 2013, 59, 208–224 CrossRef CAS PubMed.
- M. Carballa, F. Omil, J. M. Lema, M. Llompart, C. García-Jares and I. Rodríguez, et al., Water Res., 2004, 38, 2918–2926 CrossRef CAS PubMed.
- B. Kasprzyk-Hordern, R. M. Dinsdale and A. J. Guwy, Water Res., 2009, 43, 363–380 CrossRef CAS PubMed.
- L. Lishman, S. A. Smyth, K. Sarafin, S. Kleywegt, J. Toito and T. Peart, et al., Sci. Total Environ., 2006, 367, 544–558 CrossRef CAS PubMed.
- K. Fent, P. Y. Kunz, A. Zenker and M. Rapp, Mar. Environ. Res., 2010, 69, S4–S6 CrossRef CAS PubMed.
- L. L. Dobbins, S. Usenko, R. A. Brain and B. W. Brooks, Environ. Toxicol. Chem., 2009, 28, 2744–2753 CrossRef CAS PubMed.
- H. Yamamoto, I. Tamura, Y. Hirata, J. Kato, K. Kagota and S. Katsuki, Sci. Total Environ., 2011, 410, 102–111 CrossRef PubMed.
- R. U. Halden and D. H. Paull, Environ. Sci. Technol., 2005, 39, 1420–1426 CrossRef CAS PubMed.
- D. W. Kolpin, E. T. Furlong, M. T. Meyer, E. M. Thurman, S. D. Zaugg and L. B. Barber, Environ. Sci. Technol., 2002, 36, 1202–1211 CrossRef CAS PubMed.
- Y. S. Liu, G. G. Ying, A. Shareef and R. S. Kookana, Environ. Pollut., 2012, 165, 225–232 CrossRef CAS PubMed.
- S. Weiss, J. Jakobs and T. Reemtsma, Environ. Sci. Technol., 2006, 40, 7193–7199 CrossRef CAS PubMed.
- J. L. Zhao, G. G. Ying, Y. S. Liu, F. Chen, J. F. Yang and L. Wang, Environ. Toxicol. Chem., 2010, 29, 1377–1384 CAS.
- L. Staudenmaier, Ber. Dtsch. Chem. Ges., 1898, 31, 1481 CrossRef CAS.
- M. Ghavami, M. Z. Kassaee, R. Mohammadi and M. Koohi, Solid State Sci., 2014, 38, 143–149 CrossRef CAS.
- Q. Wu, G. Zhao, C. Feng, C. Wang and Z. Wang, J. Chromatogr. A, 2011, 1218, 7936–7942 CrossRef CAS PubMed.
- Y. X. Xu, H. Bai, G. W. Lu, C. Li and G. Q. Shi, J. Am. Chem. Soc., 2008, 130, 5856–5857 CrossRef CAS PubMed.
- M. Bezerraa, R. E. Santelli, E. P. Oliveira, L. S. Villar and L. A. Escaleir, Talanta, 2008, 76, 965–977 CrossRef PubMed.
- P. H. C. Eilers, I. D. Currie and M. Durban, Comput. Stat. Data Anal., 2006, 50, 61–76 CrossRef.
- R. Tauler, Chemom. Intell. Lab. Syst., 1995, 30, 133–146 CrossRef CAS.
- C. Almeida and J. M. F. Nogueira, J. Chromatogr. A, 2014, 1348, 17–26 CrossRef CAS PubMed.
- A. Prichodko, E. Janenaite, V. Smitiene and V. Vickackaite, Acta Chromatogr., 2012, 24, 589–601 CrossRef CAS.
- M. Abbasghorbani, A. Attaran and M. Payehghadr, J. Sep. Sci., 2013, 36, 311–319 CrossRef CAS PubMed.
- Y. Zhang and H. K. Lee, Anal. Chim. Acta, 2012, 750, 120–126 CrossRef CAS PubMed.
- Y. Zhang and H. K. Lee, J. Chromatogr. A, 2013, 1271, 56–61 CrossRef CAS PubMed.
- M. Payán, M. López, R. Fernández-Torres, J. Bernal and M. Mochón, Anal. Chim. Acta, 2009, 653, 184–190 CrossRef PubMed.
- Y. Ji, Z. Du, H. Zhang and Y. Zhang, Anal. Methods, 2014, 6, 7294–7304 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10675g |
| This journal is © The Royal Society of Chemistry 2016 |