
A new approach for the synthesis of sulfur-bridged polysiloxanes via thiol–ene “click” reaction and their post-functionalization to obtain luminescent materials†
Abstract
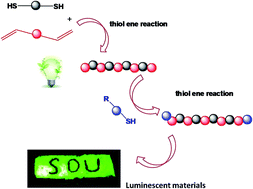
Polysiloxanes, also known as “silicones”, have received widespread attention as specialty and inorganic backbone polymers since their commercial introduction. However, traditional synthesis routes can only produce polymers with Si–O–Si bonds in the main chains; this limitation influences the rapid development of the silicone industry. As such, new methods for the synthesis of silicones must be developed. This study is the first to introduce the thiol–ene reaction as a modular, efficient, and highly orthogonal method for polymerization. Novel polysiloxanes bearing sulfur atoms in the main chain with adjustable molecular weight were synthesized by the thiol–ene “click” reaction of 1,3-divinyl-1,1,3,3-tetramethyldisiloxane. 1,2-Ethanedithiol was subsequently functionalized using the same method. The structure of these polysiloxanes was characterized by 1H NMR, 13C NMR, and GPC analyses. The new structures of polysiloxanes bearing sulfur atoms in the main chain confer novel properties and applications. Furthermore, incorporating sulfur atoms could linearly increase the refractive index of polysiloxane. Finally, hybrid luminescent materials obtained from functionalized polysiloxanes and lanthanide ions exhibited narrow-width green or red emissions.
 Please wait while we load your content...
Please wait while we load your content...

