
Surface and interface design in cocatalysts for photocatalytic water splitting and CO2 reduction
Song Bai
*ab,
Wenjie Yina,
Lili Wangb,
Zhengquan Lia and
Yujie Xiong
*b
aKey Laboratory of the Ministry of Education for Advanced Catalysis Materials, College of Chemistry and Life Sciences, Institute of Physical and Chemistry, Zhejiang Normal University, Jinhua, Zhejiang 321004, China. E-mail: songbai@zjnu.edu.cn
bHefei National Laboratory for Physical Sciences at the Microscale, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), School of Chemistry and Materials Science, University of Science and Technology of China, Hefei, Anhui 230026, China. E-mail: yjxiong@ustc.edu.cn; Fax: +86-551-63606657
First published on 3rd June 2016
Abstract
Recent advances in photocatalysis highlight the important role of cocatalysts in improving the solar-to-chemical conversion efficiency for various reactions, such as water splitting and CO2 reduction reactions. Given that cocatalysts play two important roles, in charge trapping and surface reactions, the rational material design of cocatalysts would be an effective route in pursuing their maximum contribution to the performance of photocatalysts. In this review, we aim to outline the recent progress of surface and interface design in cocatalysts for photocatalytic water splitting and CO2 reduction. We first introduce the surface design of cocatalysts, which enables the enhancement of specific water splitting or CO2 reduction reactions through surface parameter (e.g., the composition, facets and phases) adjustments. We then present key parameters for designing the interface between photocatalyst and cocatalyst, which offer a set of versatile options for tuning the charge transfer to the cocatalyst. Taken together, the surface and interface of cocatalysts may have synergetic effects on the photocatalytic performance, which are discussed to provide guidance for simultaneously tailoring surface and interface parameters. Finally, we summarize the challenges and opportunities for the surface and interface design of cocatalysts for the efficient production of solar fuels.
 Song Bai | Song Bai received his B.S. and M.S. degrees from Jiangsu University in 2009 and 2012, respectively. In 2015, he obtained his Ph.D. degree with Professor Yujie Xiong from the University of Science and Technology of China (USTC), and started as an assistant professor in Zhejiang Normal University. His research interests focus on the design and synthesis of novel hybrid nanomaterials for photocatalysis. |
 Yujie Xiong | Yujie Xiong received his B.S. in chemical physics in 2000 and his Ph.D. in inorganic chemistry in 2004 (with Professor Yi Xie), both from the University of Science and Technology of China (USTC). After four years training with Professors Younan Xia and John A. Rogers, he joined the NSF-NNIN at the Washington University in St. Louis as the Principal Scientist and Lab Manager. Starting from 2011, he is a Professor of Chemistry at the USTC. He has published 120 papers with over 10 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 citations (H-index 48). His research interests include the synthesis, fabrication and assembly of inorganic materials for energy and environmental applications.
000 citations (H-index 48). His research interests include the synthesis, fabrication and assembly of inorganic materials for energy and environmental applications.1. Introduction
Since the discovery of photocatalytic water splitting on TiO2 electrodes by Fujishima and Honda in 1972, significant efforts have been made to develop highly efficient photocatalysts for various photocatalytic reactions, such as water splitting and CO2 reduction, providing a promising route to alleviate steadily worsening environmental issues and an energy crisis.1–5 Among various photocatalyst designs, the combination of a semiconductor with a cocatalyst to form a hybrid structure is a widely used approach to promote the performance of photocatalysts in the production of solar fuels.6,7 In such photocatalytic hybrid structures, cocatalysts themselves are not the light-harvesting components for generating photoinduced charge carriers. Instead, a cocatalyst mainly plays two positive roles in steering the charge kinetics in photocatalysis: (i) trapping charge carriers to promote electron–hole separation by forming an interface with the semiconductor; and (ii) serving as a highly active reaction site to supply the trapped charges for redox reactions on their surface.8,9 Both the improved charge separation and surface catalytic reactions contribute to the enhancement of photocatalytic activity and selectivity. In addition, the use of cocatalysts as alternative reaction sites may suppress the photocorrosion of semiconductors resulting from charge carrier accumulation and thus increase the stability of photocatalysts.10The two key roles of cocatalysts highlight the importance of designing their surface and interface to maximize the improvement in photocatalyst performance.11,12 On one hand, the surface of a cocatalyst is the location for redox reactions, which greatly determines the adsorption and activation abilities for reactant molecules and thus the activity and selectivity for photocatalytic reactions.13,14 For this reason, the design of a cocatalyst surface depends on the type of chemical reaction occurring on the surface – water splitting or CO2 reduction. On the other hand, the interface between a cocatalyst and semiconductor is the location where the charge carriers are transferred and separated, holding the key to preventing adverse electron–hole recombination in the semiconductor.15
In a realistic photocatalytic system, the situation is rather complicated so as to entangle surface reactions and interfacial charge transfer, further emphasizing the significance of cocatalyst surface and interface design.11 Inefficient interfacial charge transfer would only bring a limited number of charge carriers to the surface of a co-catalyst, thereby restricting the efficacy of surface reactions. Inversely, slow surface reactions may lead to the accumulation of charges on the side of the cocatalyst, which in turn reduces the potential difference and prevents further interfacial charge transfer. In brief, only when the surface and interface are simultaneously well designed to promote surface catalytic reactions and interfacial charge transfer can the maximization of cocatalyst efficacy be realized. From another point of view, the surface and interface design of cocatalysts also represents a more straightforward strategy for improving photocatalysis in comparison with the surface modification of bare semiconductors. In the case of bare semiconductors, the semiconductor acts as both the light-harvesting center and surface reaction site. As such, tailoring the surface of the semiconductor would affect the light absorption of the photocatalyst in addition to tuning surface reactions,16 which makes it challenging to assess the contribution of surface design to photocatalytic performance.
Recently, the rational design of cocatalyst surfaces and interfaces has been widely implemented to promote the performance of photocatalytic nanomaterials. Certainly this research is greatly facilitated by the development of advanced synthetic approaches which realize design through precisely controlling surface and interface parameters of cocatalysts at the nanoscale.17,18 In this review, we focus on the design of cocatalyst surfaces and interfaces toward photocatalytic water splitting and CO2 reduction. We will first outline the forms of combination of cocatalysts with photocatalysts, as well as their basic architectural structures. Then we will discuss the surface design of cocatalysts in detail, according to the relationship between key surface parameters and photocatalysis. In the next section, the design of cocatalyst–photocatalyst interfaces will be elucidated based on a set of critical interfacial parameters. Subsequently, we will further highlight the simultaneous control of surfaces and interfaces associated with cocatalysts for enhanced photocatalytic performance. Finally, the remaining challenges and future prospects for surface and interface design in cocatalysts for photocatalytic applications will be provided.
2. Architectural structures of cocatalysts in photocatalysis
According to the trapped charge carriers that in turn determine the type of surface reaction, cocatalysts can be classified as having two functions: reduction cocatalysts trapping electrons for reduction half reactions, and oxidation cocatalysts trapping holes for oxidation half reactions. In general, noble metals (e.g., Pt, Pd, Rh and Au),19–22 non-noble transition metals (e.g., Cu, Co and Ni),23–25 metal sulfides (e.g., MoS2, NiS and WS2),26–28 metal oxides (e.g., NiO and CuO),29,30 phosphides (e.g., Co2P, NiP and MoP),31–33 and carbon materials (e.g., graphene and carbon nanotubes)34,35 can serve as reduction cocatalysts for the hydrogen evolution reaction. In parallel, noble metals (e.g., Pt, Pd and Ag),14,36,37 metal oxides (e.g., CuO, NiO and RuO2),38–40 and cobaltates (e.g., ZnCo2O4 and MnCo2O4)41,42 have been reported as reduction cocatalysts for CO2 reduction. As for oxidation cocatalysts, transitional metal oxides (e.g., IrOx, MnOx, RuOx and CoOx),43–47 and phosphates (e.g., CoP)48–50 have been widely used for water oxidation.Cocatalysts can be integrated into photocatalysis in many different forms. In a hybrid photocatalyst, reduction or oxidation cocatalysts can be loaded alone on a light-harvesting semiconductor, in which the photogenerated electrons in the conduction band (CB) or holes in the valence band (VB) of the semiconductor are transferred to the cocatalyst for a reduction or oxidation reaction (Fig. 1a and b).14,48 In other cases, reduction and oxidation cocatalysts can be co-loaded on the same light-harvesting semiconductor for reduction and oxidation reactions (Fig. 1c).51,52 It is worth pointing out that the semiconductor for loading cocatalysts is not necessarily photoexcited. Alternatively, plasmonic metals or photosensitizers are integrated with a semiconductor so that the photoexcited metals or sensitizers can inject hot electrons or holes into the CB or VB of the semiconductor, respectively. As such, the injected electrons or holes are further transferred to the reduction or oxidation cocatalyst for surface reactions (Fig. 1d).53–55 Certainly in rare circumstances, cocatalysts may directly interface with plasmonic metals or photosensitizers to form hybrid photocatalysts in the absence of a semiconductor (Fig. 1e).56–59 The hot charge carriers generated in the plasmonic metal or photosensitizer are directly injected to the cocatalyst for redox reactions without the bridge of a semiconductor.
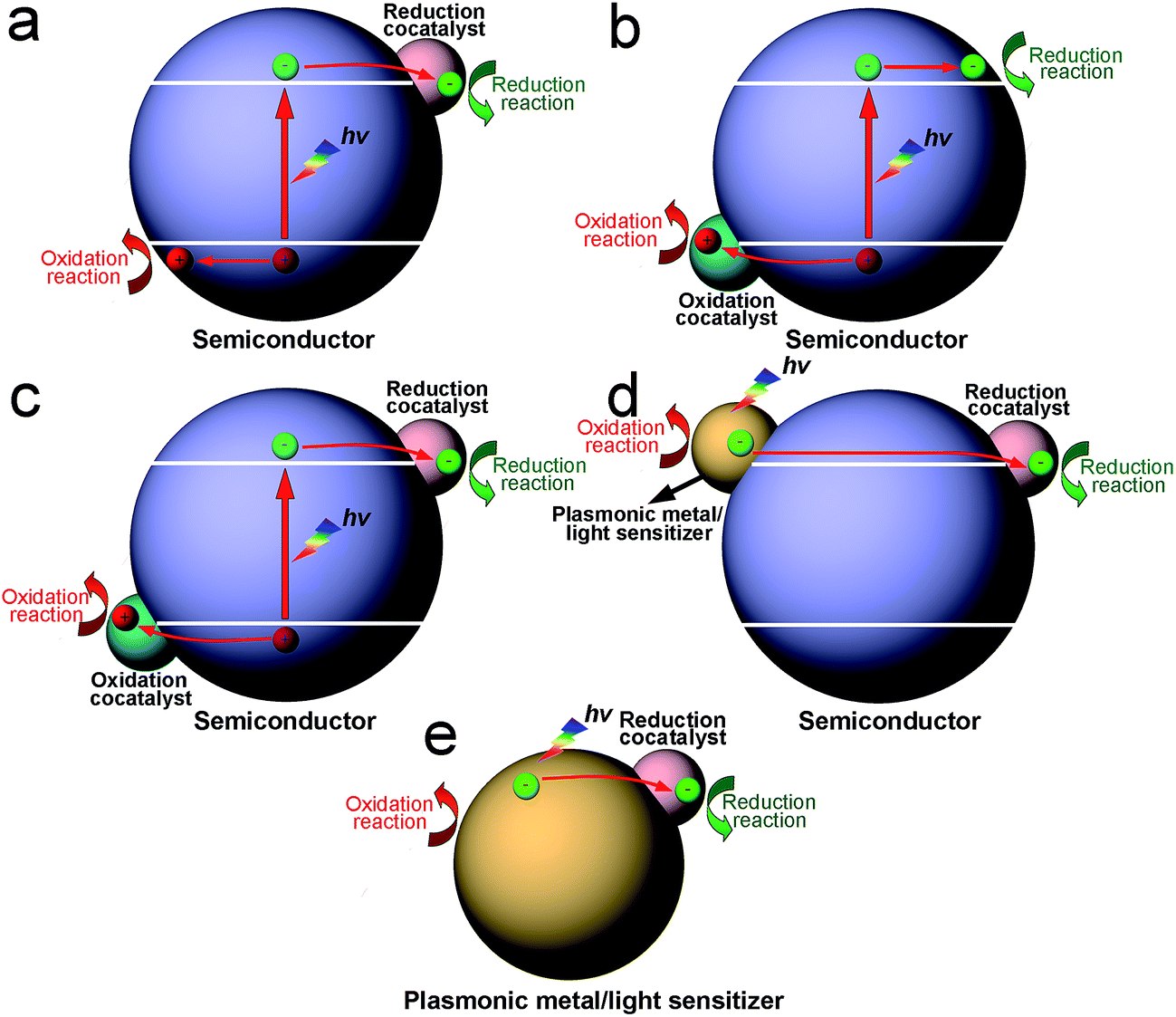 | ||
| Fig. 1 Schematic diagram illustrating the integration of photocatalysts with cocatalysts in various configurations: (a) semiconductor–reduction cocatalyst structure; (b) semiconductor–oxidation cocatalyst structure; (c) reduction cocatalyst–semiconductor–oxidation cocatalyst structure; (d) plasmonic metal/sensitizer–semiconductor–cocatalyst structure; and (e) plasmonic metal/sensitizer–cocatalyst structure. | ||
As the viewpoint is further narrowed down to a single reduction or oxidation cocatalyst, the integration of a cocatalyst with a semiconductor can still involve a variety of basic architectural structures, particularly in the case of reduction cocatalysts. This situation involves many different surface and interface structures in the cocatalyst design. To simplify the case, here we mainly take reduction cocatalysts as examples. When the cocatalyst is a mono-component structure (namely, a semiconductor–cocatalyst I structure), the cocatalyst surface and semiconductor–cocatalyst interface are quite simple (Fig. 2a).14,26,37,60–62 As the reduction cocatalyst has two or more components involved, the models for integrating the components become more diversified. In one case, two reduction cocatalysts (namely, cocatalyst I and cocatalyst II) in mutual contact are loaded on the same semiconductor. In this so-called “semiconductor–cocatalyst I/II structure”, two semiconductor–cocatalyst interfaces (i.e., the interfaces of semiconductor–cocatalyst I and semiconductor–cocatalyst II) as well as a cocatalyst I–cocatalyst II interface are simultaneously formed for electron transfer, and meanwhile, the surfaces of the two cocatalysts are both exposed for reduction reactions (Fig. 2b).63,64 In another case, a quasi-core–shell structure is formed between cocatalyst I and cocatalyst II, fabricated by selectively coating the semiconductor-supported cocatalyst I core with a shell of cocatalyst II (i.e., a semiconductor–cocatalyst I@II core–shell structure) (Fig. 2c).21,65–67 In this structure, the interfaces of semiconductor–cocatalyst I and cocatalyst I–cocatalyst II are formed for tandem electron transfer, leaving only the cocatalyst II surface exposed for reduction reactions. Similarly, the semiconductor–cocatalyst I–II structure, in which cocatalyst II is selectively loaded on cocatalyst I to form a supported structure (Fig. 2d),68,69 has both the interfaces of semiconductor–cocatalyst I and cocatalyst I–cocatalyst II designed for electron transfer. In this case, however, both the surfaces of cocatalyst I and cocatalyst II are exposed for reduction reactions.
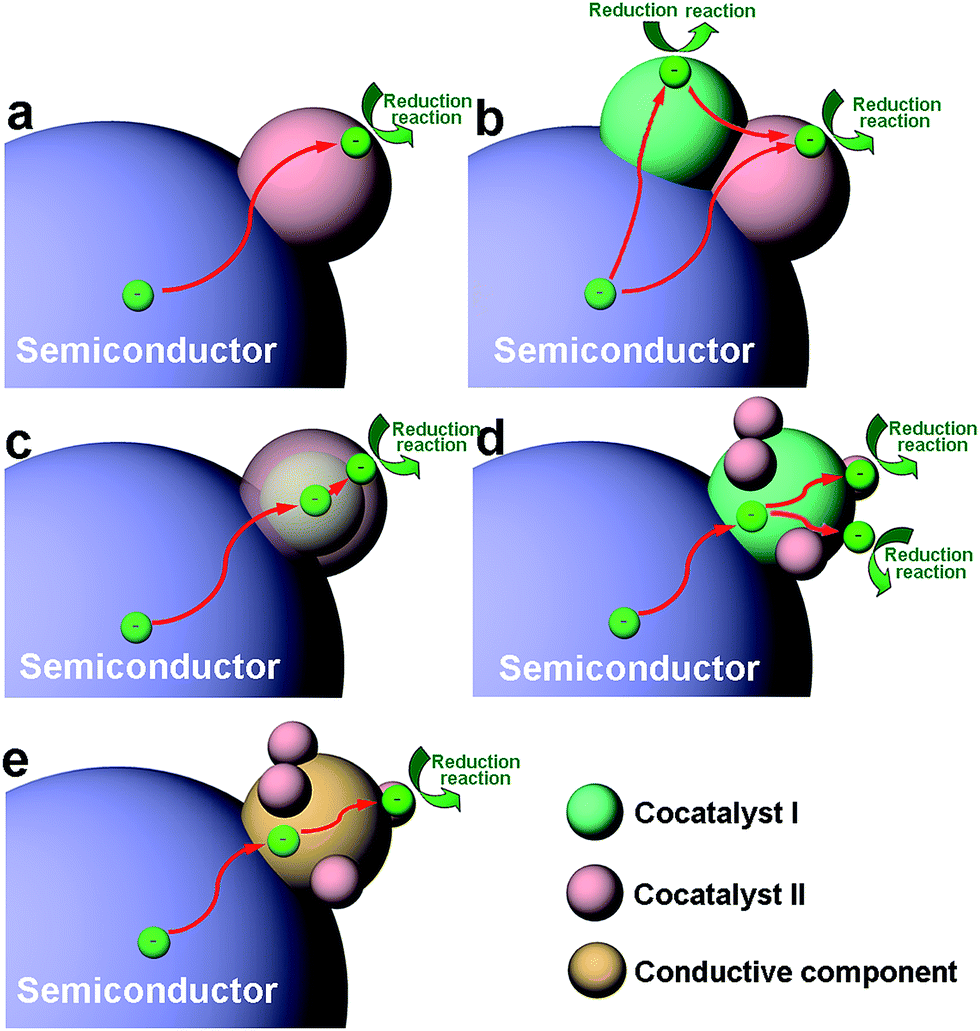 | ||
| Fig. 2 Schematic diagram illustrating the architectural structures of cocatalysts for photocatalysis, as well as the functions of their surfaces and interfaces for photocatalytic reactions: (a) semiconductor–cocatalyst I structure; (b) semiconductor–cocatalyst I/II structure; (c) semiconductor–cocatalyst I@II core–shell structure; (d) semiconductor–cocatalyst I–II structure; and (e) semiconductor–conductor–cocatalyst structure. Here a reduction cocatalyst is used as an example. | ||
Certainly it is not necessary to have the cocatalyst in direct contact with the semiconductor. Alternatively, a conductive component (e.g., graphene or carbon quantum dots) can also serve as a charge bridge between the cocatalyst and semiconductor to form a semiconductor–conductor–cocatalyst structure (Fig. 2e).70–72 As the conductor surface barely possesses catalytic activity, a tandem electron transfer through the interfaces of semiconductor–conductive layer and conductive layer–cocatalyst will designate the cocatalyst surface as the reduction reaction site. In the literature, there have been reported more complicated architectural structures for cocatalysts (e.g., ternary cocatalysts), and they still derive from one of the aforementioned structures or the combination of multiple structures.73 From the discussion above, it can be recognized that the mutual interfaces of the cocatalyst–cocatalyst or cocatalyst–conductor are also of great importance to the charge transfer in photocatalysis when multiple components are involved in the cocatalyst design.
3. Surface design of cocatalysts in photocatalysis
The analysis above clearly reveals that the cocatalyst surface is the location where activation reactions take place during the photocatalytic process. For this reason, surface design holds promise for tuning photocatalytic reactions through tailoring some surface parameters of cocatalysts. Firstly, surface parameters can be designed not only to realize high adsorption and activation ability for specific reactant molecules, but also to prevent side or back reactions. This would enhance the activity in the main photocatalytic reaction as well as improve the selectivity between competing reactions. Secondly, surface parameters are critical for the accumulation of electrons or holes on the reactive surface for reduction or oxidation reactions, respectively. Thirdly, the chemical stability of photocatalysts during the catalytic process can be improved by modifying the surface parameters. In this section, the surface design of cocatalysts for photocatalytic H2 evolution and CO2 reduction is discussed according to some important surface parameters such as surface composition, facets, phases and defects.3.1 Surface composition
The surface composition greatly determines the atomic or ionic arrangements on a cocatalyst surface and thus the adsorption and activation behavior of reactants. To date, the surface design of cocatalysts through composition optimization has been widely used to enhance the performance of photocatalysts for various reactions. As the adsorption and activation behaviors are the keys to this performance tuning, the selection of cocatalyst compositions certainly depends on the type of reaction – water splitting or CO2 reduction. For instance, metallic Pt is the most widely used reduction cocatalyst in water splitting mainly due to its low activation energy for H2 evolution.6 However, the back reaction of hydrogen oxidation may also be catalyzed by the metallic Pt cocatalyst, thus limiting the solar energy conversion efficiency. To solve this problem, Li et al. loaded Pt in an oxidized state (PtO) on anatase TiO2 nanosheets (Fig. 3a), using a similar chemical reduction method to metallic Pt, except for the addition of a poly(methacrylic acid) ligand.74 Fig. 3b summarizes the photocatalytic performance of the two samples in pure water splitting, with the pre-injection of stoichiometric H2 and O2 into the closed test system. The TiO2–Pt photocatalyst exhibited a remarkable decrease in H2 and O2 amounts with extended periods of light irradiation, mainly resulting from the undesirable hydrogen oxidation reaction (HOR) on the Pt cocatalyst. In sharp contrast, the TiO2–PtO offered the capability for stoichiometric H2 and O2 to evolve steadily as the reaction proceeded. The cocatalyst PtO not only acts as efficient H2 evolution sites, but also exhibits the remarkable ability of suppressing the HOR as it can hardly activate and dissociate H2 molecules (Fig. 3c).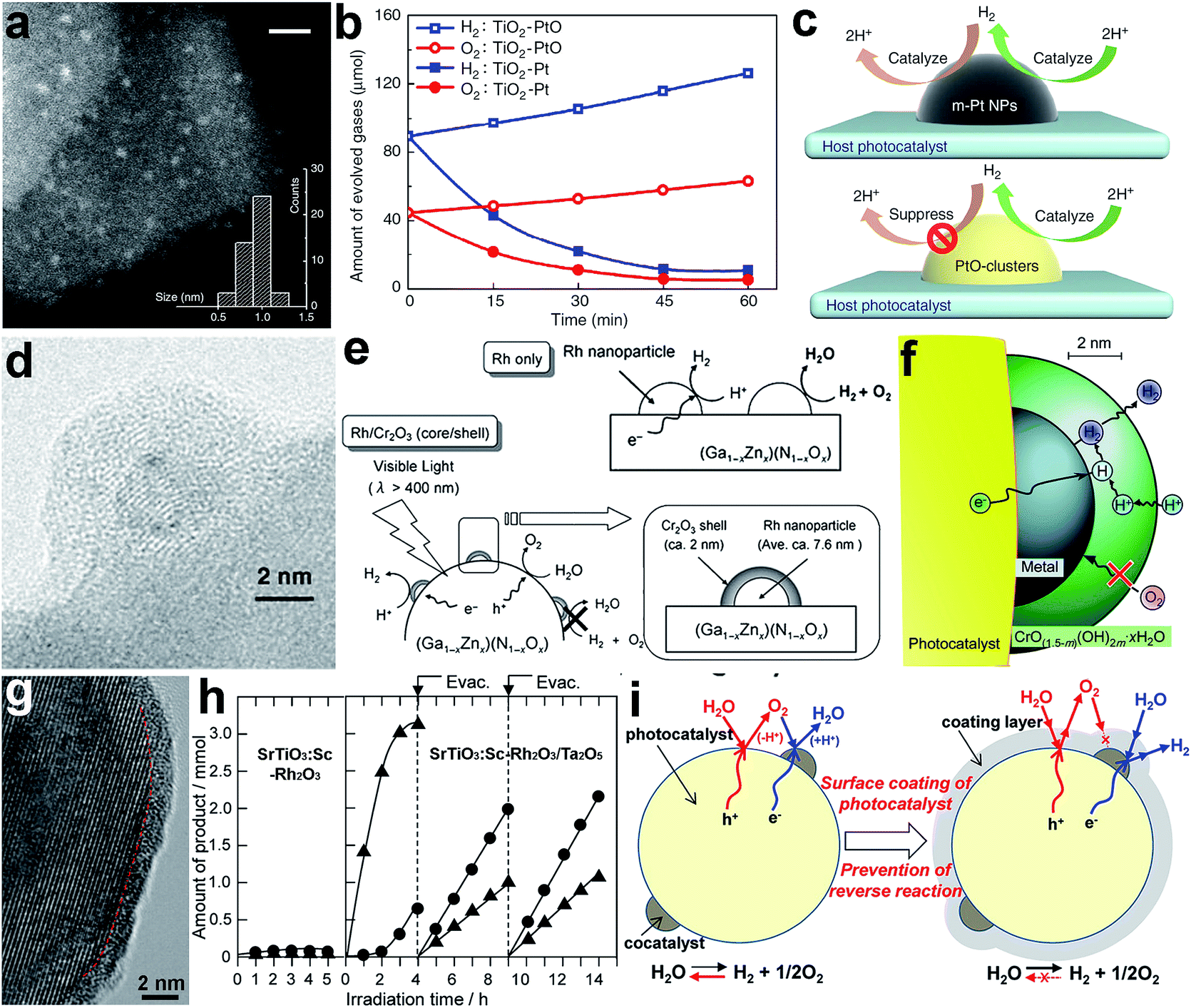 | ||
| Fig. 3 (a) STEM image of a TiO2–PtO photocatalyst, (b) time-resolved profiles of the HOR with pre-injected H2 and O2 on TiO2–Pt and TiO2–PtO photocatalysts under light irradiation, (c) schematic diagram illustrating the surface reactions on Pt and PtO cocatalysts during water splitting (adapted with permission from ref. 74, Copyright 2013 Nature Publishing Group); (d) TEM image of (Ga1−xZnx)(N1−xOx)-supported Rh@Cr2O3 core–shell cocatalyst; (e) schematic diagram illustrating the surface reactions on Rh and Rh@Cr2O3 core–shell cocatalysts during water splitting; (f) mechanism of Rh@Cr2O3 core–shell cocatalysts for water splitting (adapted with permission from ref. 21 and 75, Copyright 2006 Wiley-VCH and 2009 American Chemical Society); (g) HRTEM image of Ta2O5-coated SrTiO3:Sc–Rh2O3 photocatalyst; (h) time course of gas evolution during the photoirradiation of pure water with SrTiO3:Sc–Rh2O3 and SrTiO3:Sc–Rh2O3/Ta2O5 as the photocatalyst; (i) schematic diagram illustrating the reaction mechanism for overall water splitting on SrTiO3:Sc–Rh2O3 and SrTiO3:Sc–Rh2O3/Ta2O5 core–shell photocatalysts (adapted with permission from ref. 84, Copyright 2015 American Chemical Society). | ||
In photocatalytic water splitting, another effective strategy for suppressing the back reaction of hydrogen oxidation on noble metals is to develop core–shell cocatalysts. In a typical case, to prevent the HOR on a metal cocatalyst, Cr2O3 was selectively coated on the noble metal (e.g., Rh) to form an M@Cr2O3 core–shell cocatalyst. The Cr2O3 surface provided alternative H2 evolution sites and prevented the back reaction, while the metal facilitated electron migration toward the Cr2O3 surface (Fig. 3d and e).21 In a further study, a selective permeation mechanism was proposed for the core–shell cocatalyst, in which the Cr2O3 layer (i.e., microporous CrO(1.5−m)(OH)2m·xH2O in aqueous solution) does not interfere with proton reduction at the Cr2O3–Pt interface and the diffusion of H2, but suppresses the permeation of oxygen atoms and molecules (Fig. 3f).75 Later on, metal@Cr2O3 core–shell cocatalysts have been widely used in photocatalysis and further extended to metal oxide@Cr2O3 core–shell cocatalysts.67,76–78 With a similar mechanism, the Ni@NiO core–shell structure is another important cocatalyst with the capability of inhibiting the back reaction in water splitting.18,79–81 In the structure, the NiO shell allows the diffusion of protons to reach Ni sites for reduction reactions as well as the diffusion and escape of the produced H2, but it can act as a valid barrier for O diffusion.
As a matter of fact, Cr2O3 has been extensively employed as a second cocatalyst component in UV-excitable photocatalysis with wide-bandgap semiconductors. Recently, novel surface modification methods have offered alternative materials to replace the Cr2O3 shell to achieve overall water splitting.82–84 A layer of amorphous transition-metal oxynitride or oxyhydroxide covered the entire surface of the semiconductor and cocatalyst, and functioned as a molecular sieve to selectively filter reactant and product molecules. For instance, to prevent the backward oxygen reduction reaction (ORR) reaction on the surface of a SrTiO3-supported Rh2O3 cocatalyst, oxyhydroxide layers of Ta5+ (referred to as Ta2O5 for simplicity) were formed to fully cover the photocatalyst to form SrTiO3:Sc–Rh2O3/Ta2O5 core–shell structures (Fig. 3g).84 In the absence of a Ta2O5 coating, the photocatalytic H2 and O2 evolution on SrTiO3:Sc–Rh2O3 was largely limited by the rapid backward reaction (Fig. 3h). In comparison, SrTiO3:Sc–Rh2O3/Ta2O5 behaved very differently. Upon irradiation, a considerable amount of O2 evolved during the initial 3–4 h due to contaminants from the catalyst synthesis. After evacuating the reaction system, H2 and O2 were produced at constant rates and at the stoichiometric ratio of water splitting. It was demonstrated that the ORR back reaction was successfully prevented by the Ta2O5 coating. In this design, the amorphous Ta2O5 layer allows H+ ions and H2O molecules to reach the surface of Rh2O3 and SrTiO3 for H2 and O2 evolution, respectively. Although the produced O2 can be released from the coating layer through penetration, O2 permeation in the opposite direction is unlikely to occur as the partial O2 pressure in the outer phase is lower than in the coating layer. Such one-way permeation of O2 effectively prevents the back reaction without compromising the forward reaction (Fig. 3i).
In addition to water splitting, the surface composition also plays an important role in the photocatalytic reduction of CO2 with H2O. For instance, a Pt@Cu2O core–shell cocatalyst was designed to enhance photocatalytic selectivity in the reduction of CO2 to CO and CH4.13 Given the high ability of the Pt cocatalyst for H2O activation, H2 would be the major product when a TiO2–Pt hybrid structure is used as a photocatalyst, reducing the selectivity of CO2 conversion. To improve the selectivity, Cu2O, a material with high CO2 activation ability, was selectively coated on the Pt to modify the surface composition (Fig. 4a). As shown in Fig. 4b, the Cu2O coating significantly suppressed the formation of H2 and promoted the production of CH4 and CO, remarkably increasing the selectivity for CO2 reduction. In this structure, the Pt core transferred the photogenerated electrons from TiO2 to the Cu2O shell, and the Cu2O shell served as the reaction sites for producing CH4 and CO (Fig. 4c).
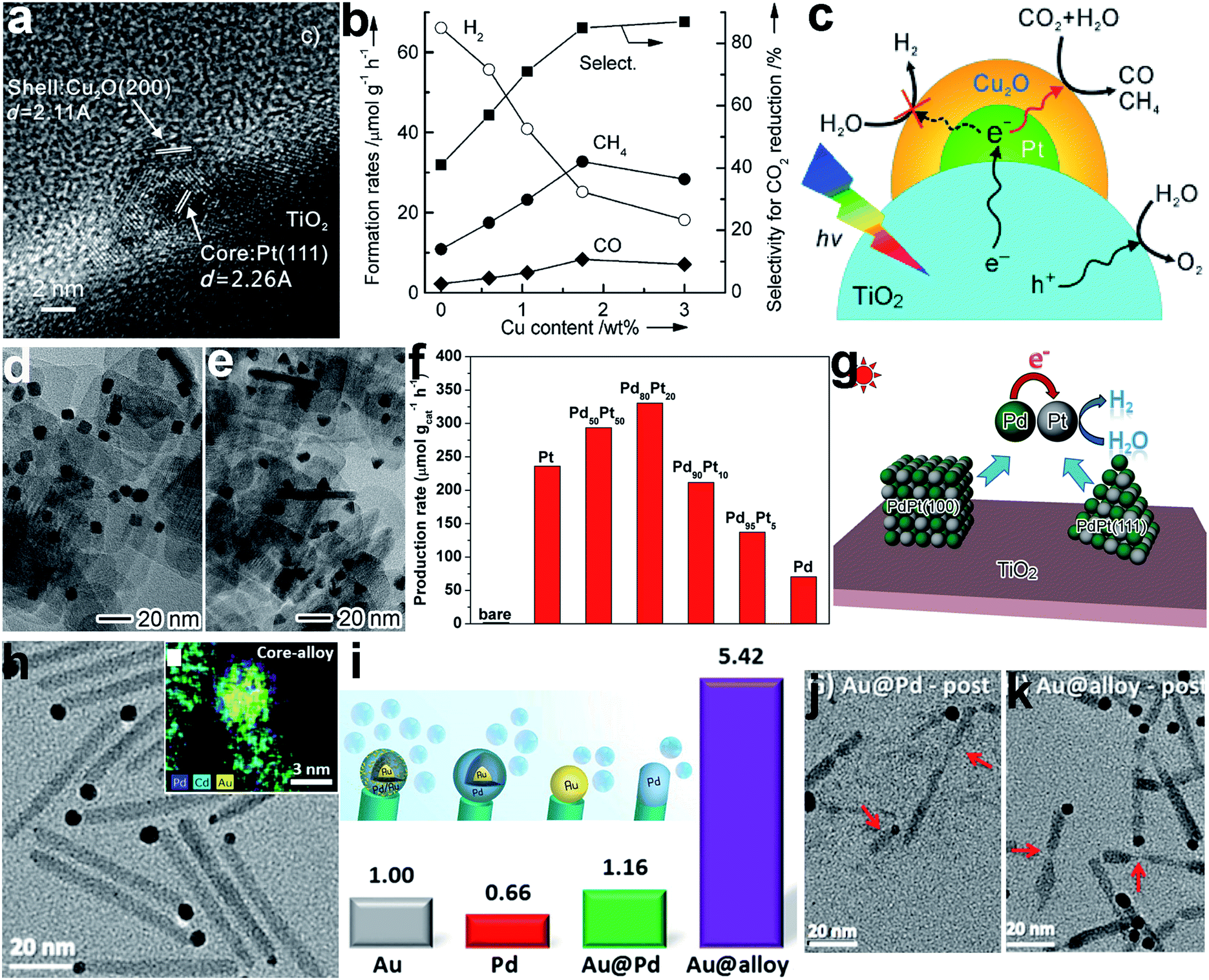 | ||
| Fig. 4 (a) HRTEM image of a TiO2-supported Pt@Cu2O core–shell cocatalyst, (b) the dependence of photocatalytic behavior on Cu content in TiO2–Pt@Cu2O photocatalysts for the reduction of CO2 with H2O, (c) a schematic illustration of the selective coating of a Pt cocatalyst with Cu2O to enhance photocatalytic CO2 reduction selectivity (adapted with permission from ref. 13, Copyright 2013 Wiley-VCH); (d and e) TEM images of TiO2-supported PdPt alloy (d) nanocube and (e) nanotetrahedron cocatalysts, (f) hydrogen production rates using TiO2-supported Pd and Pt and PdPt alloy (with different Pd/Pt ratios) nanocube cocatalysts, (g) schematic illustration of photocatalytic H2 evolution on TiO2-supported PdPt alloy cocatalysts (adapted with permission from ref. 86, Copyright 2016 Wiley-VCH); (h) TEM image and EDS elemental mapping of CdSe@CdS rods with Au@PdAu alloy core–shell cocatalysts on their tips, (i) schematic illustration and relative hydrogen production rates of CdSe@CdS seeded rods with Pd, Au@Pd, and Au@(Au/Pd alloy) tips serving as reduction cocatalysts relative to their Au-tip counterpart, and (j and k) TEM images of CdSe@CdS with (j) Au@Pd and (k) Au@(Au/Pd alloy) tips after photocatalysis (adapted with permission from ref. 87, Copyright 2015 American Chemical Society). | ||
In addition to surface coating or decoration, surface composition control can also be achieved through incorporating new atoms into a cocatalyst surface. Metal cocatalysts can be tailored by forming alloys with different metal atoms.85,86 For instance, H2 evolution using TiO2 nanosheets with a Pt cocatalyst could be enhanced by incorporating Pd into the Pt lattice to form a PdPt alloy cocatalyst with both cases of Pt/Pd{100} and {111} surface facets at various typical Pd/Pt ratios (Fig. 4d–f).86 In this system, the difference in the work functions of Pd and Pt induced electron accumulation at Pt sites. The increase in electron density in turn enhanced the H2O adsorption and activation on the Pt sites for H2 production (Fig. 4g). In another case, an Au@(AuPd alloy) core–shell cocatalyst has been developed for efficient H2 production with CdSe@CdS as the photoactive charge generation unit (Fig. 4h).87 The CdSe@CdS rods with Au@alloy core–shell tips exhibited a significant enhancement in photocatalytic activity in comparison with Pd, Au and Au@Pd core–shell tips, benefiting from both the alteration in electronic structure by the Au core and the atomic rearrangement of the Pd surface (Fig. 4i). In the AuPd alloy, Pd reaction sites are separated with Au, which reduces the H adsorption strength to release H2 as well as eliminating the so-called self-poisoning effect. Furthermore, the Au on the surface enhances the photocatalytic stability by suppressing cation exchange reactions between Cd and Pd (Fig. 4j and k).
3.2 Surface facets
With the same surface composition, tailoring the facets exposed on a cocatalyst surface can also lead to a variation in atomic arrangements. Similarly, this would maneuver the adsorption and activation of reactive molecules, and tune the photocatalytic activity and selectivity. During the process of photocatalytic conversion of CO2 and H2O into carbon fuels, water splitting often competes with CO2 reduction by consuming photoexcited electrons. In a typical case, the selectivity between CO2 reduction and water splitting can be tuned through adjusting the exposed facets of Pd reduction cocatalysts supported on g-C3N4 nanosheets.14 In this example, Pd nanocubes enclosed with {100} facets and Pd nanotetrahedrons with {111} facets were grown in situ on C3N4 nanosheets to form C3N4–Pd{100} and C3N4–Pd{111} photocatalysts, respectively (Fig. 5a and b). With the same Pd loading (ca. 6 wt%), C3N4–Pd{100} preferred to reduce H2O to H2, while C3N4–Pd{111} mainly supported CO2 reduction to carbon products (i.e., CO, CH4 and C2H5OH). As a result, the selectivity for CO2 reduction turned out to be 20.7% for Pd{100} versus 78.1% for Pd{111} (Fig. 5c). Behind the observations, theoretical simulations revealed that the Pd{111} facets offered higher CO2 adsorption energy and a lower CO2 activation barrier, while the Pd{100} facets possessed much higher H2O adsorption energy. Despite comparable electron transfer efficiencies, the electrons trapped on various facets of the Pd cocatalyst would be mainly utilized for different reduction reactions (Fig. 5d). Besides Pd, the exposed facets of Pt cocatalysts have also been reported to play an important role in determining the photocatalytic performance for H2 production.88,89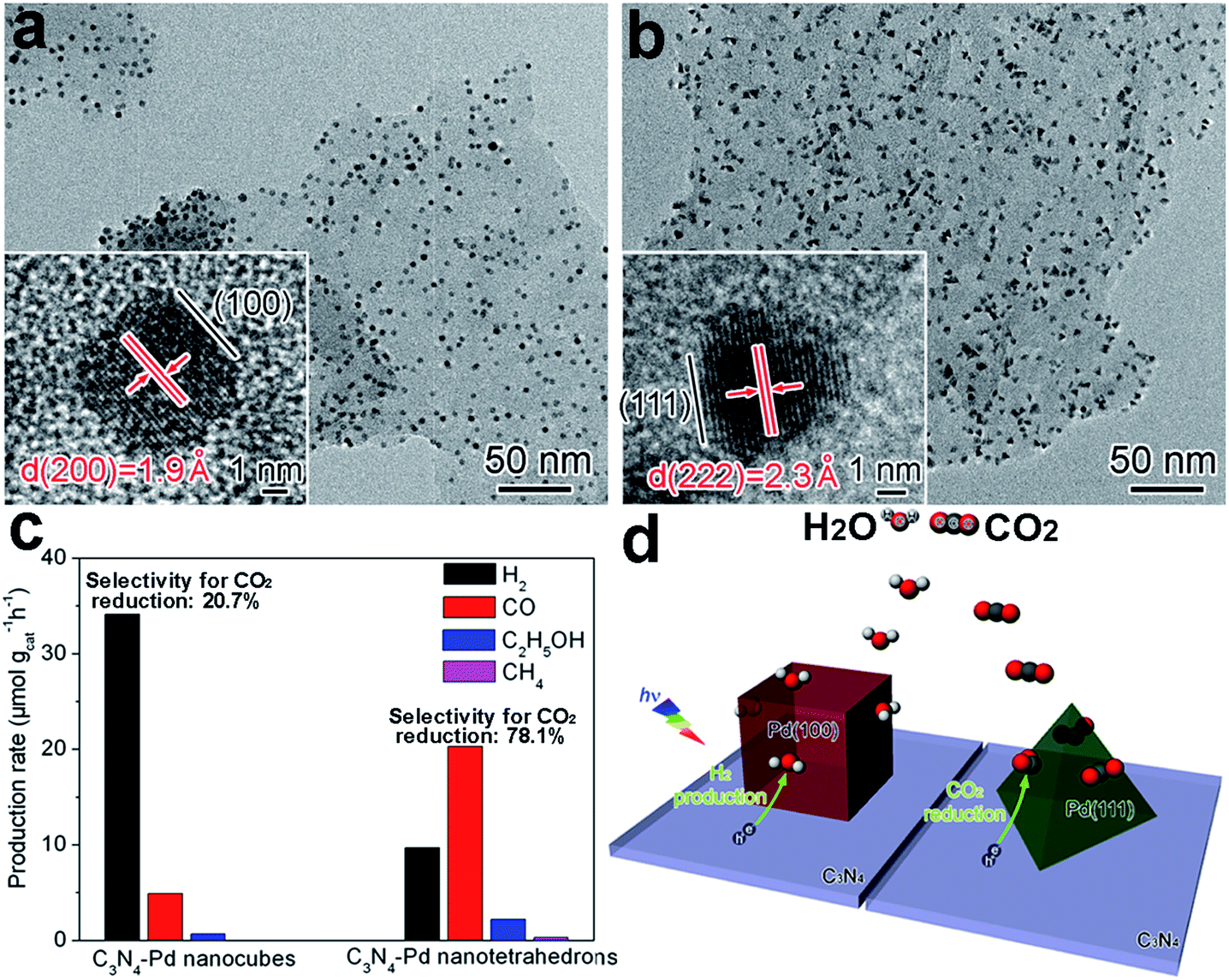 | ||
| Fig. 5 (a and b) TEM and HRTEM images of (a) C3N4–Pd nanocubes with exposed Pd{100} facets and (b) C3N4–Pd nanotetrahedrons with Pd{111} facets; (c) production rates of H2 and carbon products using C3N4–Pd photocatalysts with ca. 6 wt% Pd loading; and (d) schematic illustration of C3N4–Pd{100} and C3N4–Pd{111} samples in photocatalytic CO2 reduction in the presence of H2O (adapted with permission from ref. 14, Copyright 2014 Royal Society of Chemistry). | ||
3.3 Surface phase
When we design the surface of a cocatalyst, the surface phase is another important parameter that has to be taken into account. For instance, it turned out that different photocatalytic H2 production rates could be achieved when metallic (1T, octahedral phase) and semiconducting (2H, trigonal prismatic phase) MoS2 were used as reduction cocatalysts with light-harvesting TiO2.90 In this case, comparable TiO2 nanocrystals were loaded on the MoS2 nanosheets in 1T and 2H phases to form TiO2–MoS2(1T) and TiO2–MoS2(2H) hybrid structures (Fig. 6a). As shown in Fig. 6b, TiO2–MoS2(1T) exhibited a dramatically higher photocatalytic H2 production rate in comparison to TiO2–MoS2(2H). The advantages of the 1T phase in cocatalysts originated from two aspects: (i) the active sites for H2 evolution were only located at the edges of the 2H MoS2 nanosheets, while the 1T MoS2 nanosheets had abundant active sites at both the edges and basal planes; and (ii) the 1T MoS2 nanosheets offered significantly higher mobility for electron transfer in comparison with the 2H MoS2 nanosheets. As such, the higher diffusion rate and shorter diffusion distance ensure more photogenerated electrons from TiO2 arrive at the reaction sites of the 1T MoS2 cocatalyst and participate in photocatalytic reactions (Fig. 6c). Owing to these advantages, transition metal oxides and phosphides (e.g., MoO2 and MoP) with surface metallic phases have also been developed as highly efficient noble-metal-free cocatalysts for photocatalytic hydrogen production from water.32,60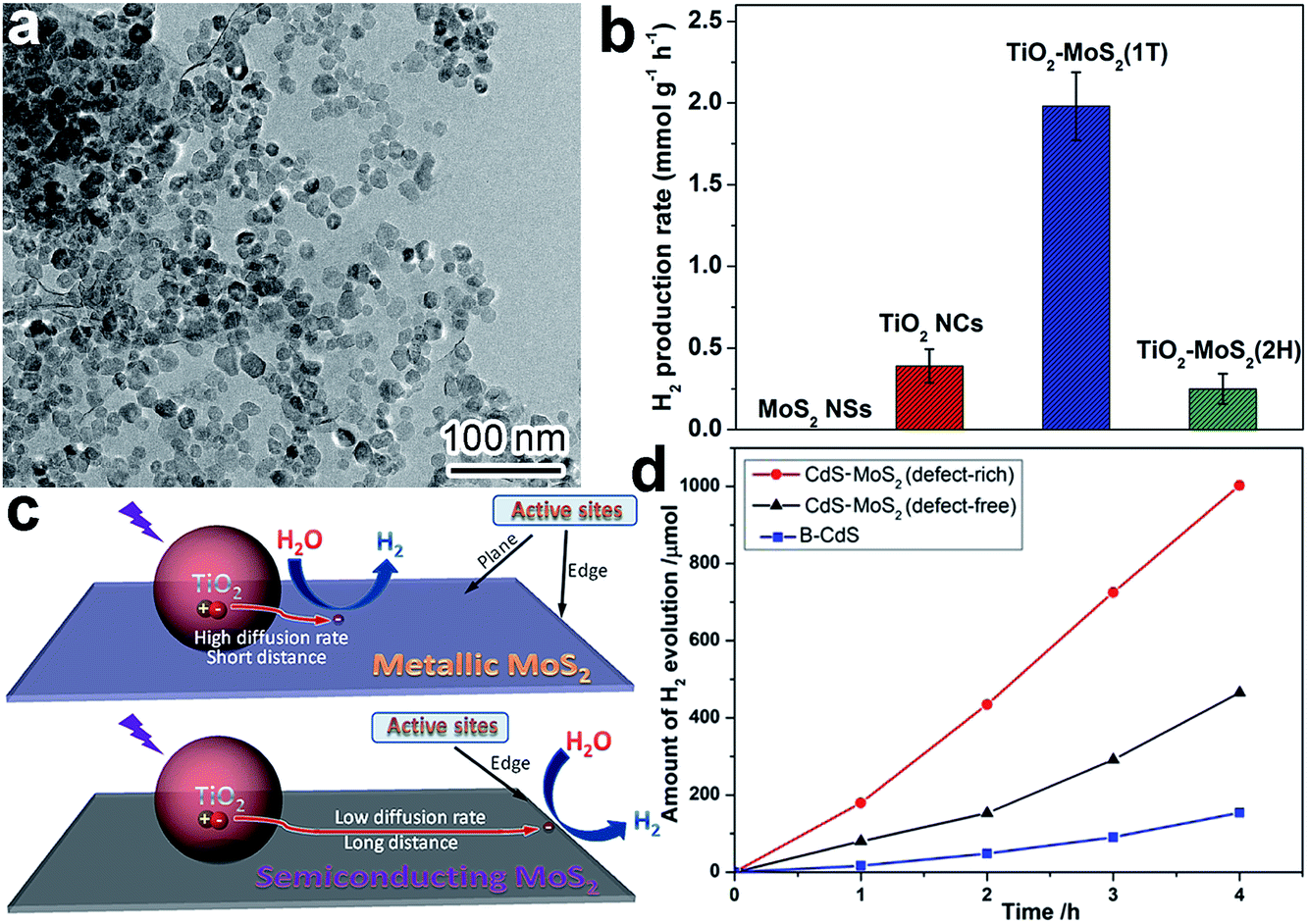 | ||
| Fig. 6 (a) TEM image of TiO2–MoS2(1T) photocatalyst; (b) photocatalytic hydrogen production rates of TiO2–MoS2; (c) schematic diagram illustrating charge-transfer behavior and H2 evolution active sites on TiO2–MoS2(1T) and TiO2–MoS2(2H) (adapted with permission from ref. 90, Copyright 2015 Springer); and (d) time course of H2 evolution over CdS, CdS–MoS2 (defect-free) and CdS–MoS2 (defect-rich) (adapted with permission from ref. 91, Copyright 2015 Royal Society of Chemistry). | ||
3.4 Surface defects
In terms of surface photocatalytic reactions, the adsorption and activation of reactants often take place at surface defects or vacancies where dangling bonds are prone to capture both charge carriers and reactants. Thus the activity and selectivity in photocatalysis can be improved by controlling the number of surface defects or altering the type of surface defects. For instance, CdS nanocrystals were anchored on defect-free and defect-rich MoS2 ultrathin nanoplates, respectively, to act as H2 evolution cocatalysts.91 The CdS–MoS2 (defect-rich) structure exhibited dramatically higher H2 evolution activity compared to CdS–MoS2 (defect-free), mainly owing to the role of defects as reactive sites (Fig. 6d). It should be noted that here we do not bother to consider the side effects of surface defects on charge recombination, as only electrons or holes are trapped on the surface of cocatalysts.4. Interface design of cocatalysts for photocatalysis
Differently from surface design, interface design is mainly performed via parameter adjustments and optimization of the contact interfaces between cocatalysts and other components. The interface has to be tightly controlled simply because it is the location through which the photogenerated charge carriers are transferred. As a matter of fact, interface design enables an improvement in photocatalytic performance from several different angles. Firstly, interface parameters can be designed to realize highly efficient charge transfer. The efficiency of charge transfer determines the number of charge carriers for redox reactions at the cocatalyst surface. Secondly, spatial charge separation can be enabled through interface control to prevent detrimental electron–hole recombinations in the semiconductor. Thirdly, the interface quality actually represents the bonding between the cocatalyst and other components, so the stability of the photocatalyst is largely relevant to the interface. In this section, we will discuss interface design according to key interface parameters, including interfacial composition, location and facets.4.1 Interfacial composition
Similarly to surface composition, the composition of the interface, where the cocatalyst contacts with the adjacent component, is a key parameter for affecting the efficiency of charge transfer across the interface. For instance, the similar composition of the two sides of an interface would favor their intimate contact and strong coupling for highly efficient charge transfer. In typical cases, transition metal sulfides (e.g., MoS2, WS2 and NiS) were often used as H2 evolution cocatalysts for CdS-based photocatalysts,26–28,91 because their analogous compositions (i.e., containing S2− anions) favored the formation of a covalent junction with a low defect density at the interface to facilitate electron transfer.Recently, interfacial composition adjustment on the cocatalyst has been reported to improve the performance of photocatalysts. For instance, the interface between a CoOx oxidation co-catalyst and a Ta3N5 semiconductor has been tailored toward improved water oxidation.15 As intimate contact could hardly be made between the hydrophobic Ta3N5 and hydrophilic CoOx for efficient interfacial charge transfer, a magnesia nanolayer was used to turn the Ta3N5 surface from hydrophobic to hydrophilic through in situ or ex situ surface coating prior to CoOx deposition, forming Ta3N5–(in)MgO/CoOx and Ta3N5–(ex)MgO/CoOx, respectively (Fig. 7a). The magnesia coating not only improved the interfacial contact between CoOx and Ta3N5, but also reduced the defect density of Ta3N5 through a passivation effect (Fig. 7b). As a result, Ta3N5–(in)MgO/CoOx and Ta3N5–(ex)MgO/CoOx exhibit substantially higher oxygen evolution rates than Ta3N5–CoOx (Fig. 7c). The transient absorption spectra revealed that the MgO layer effectively suppressed the recombination of photoinduced carriers and prolonged their lifetimes (Fig. 7d).
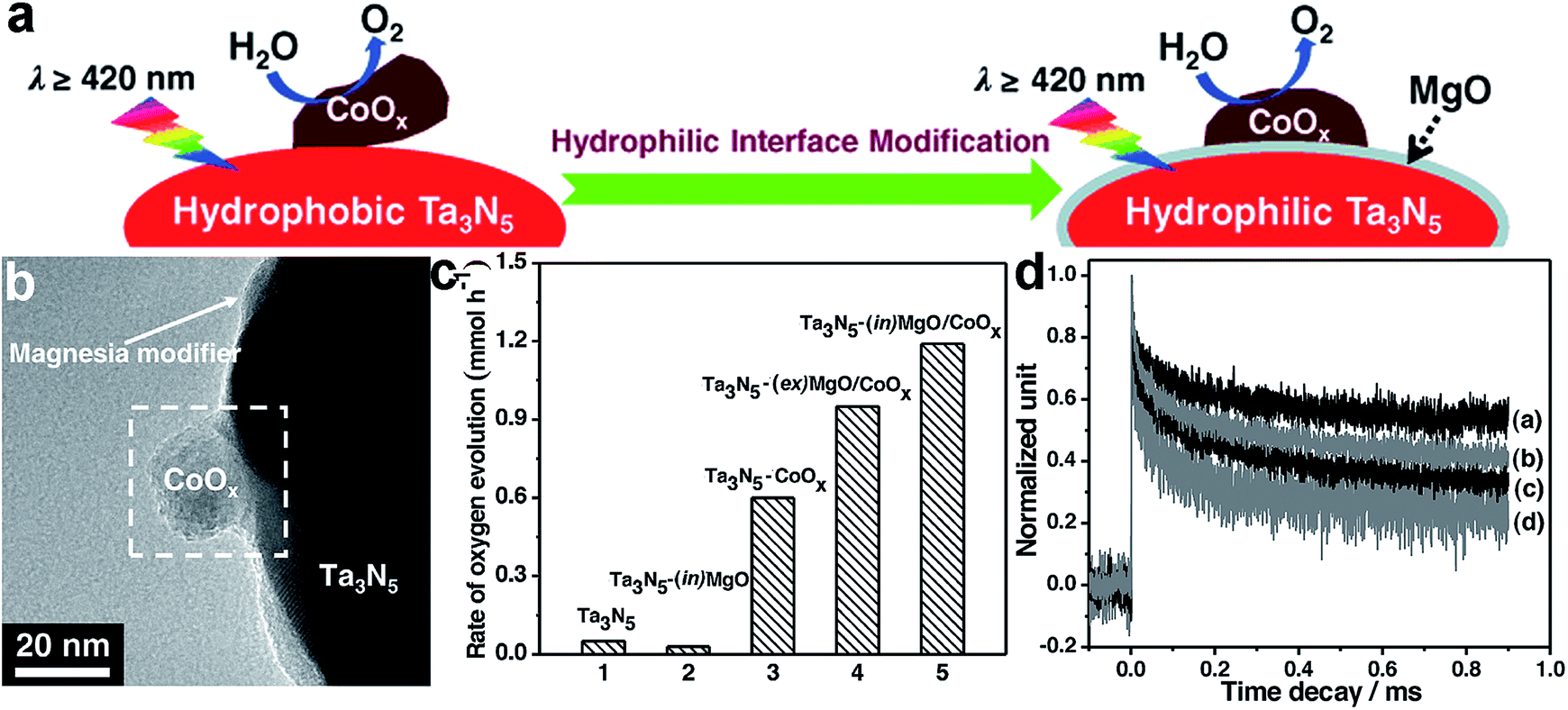 | ||
| Fig. 7 (a) Schematic diagram illustrating hydrophilic interface modification for improving the interfacial contact between Ta3N5 and CoOx for enhanced water oxidation; (b) TEM image of Ta3N5–(in)MgO/CoOx; (c) rates of oxygen evolution on the Ta3N5-based photocatalysts; and (d) the decay in transient absorption for the representative Ta3N5-based photocatalysts: (a) Ta3N5–(in)MgO/CoOx, (b) Ta3N5–(ex)MgO/CoOx, (c) Ta3N5–CoOx and (d) Ta3N5 (adapted with permission from ref. 15, Copyright 2015 Wiley-VCH). | ||
4.2 Interfacial location
The location for forming the interface is critical to the efficiency of interfacial charge transfer, especially when electrons and holes are accumulated on different components in a heterostructure. To ensure high charge-transfer efficiency, reduction or oxidation cocatalysts should be deposited on locations where electrons or holes are accumulated, respectively. For instance, Park et al. reported that the configuration of loading Pt cocatalysts onto CdS/TiO2 hybrid catalysts greatly determined the overall H2 production efficiency.92 The Pt cocatalyst could be deposited on the surface of either CdS (i.e., TiO2/CdS–Pt) or TiO2 (i.e., CdS/TiO2–Pt) or on both surfaces, to form different interface configurations (Fig. 8a). Under visible light, the CdS/TiO2–Pt structures exhibited remarkably higher H2 production rates in comparison with (CdS/TiO2)–Pt and TiO2/CdS–Pt (Fig. 8b and c), as the decoration of Pt cocatalyst on the TiO2 side enabled successive electron transfer along CdS → TiO2 → Pt and maximized the electron transfer efficiency. In a different way, the multi-directional electron transfer throughout (CdS/TiO2)–Pt and TiO2/CdS–Pt inevitably lowered the efficiency (Fig. 8a).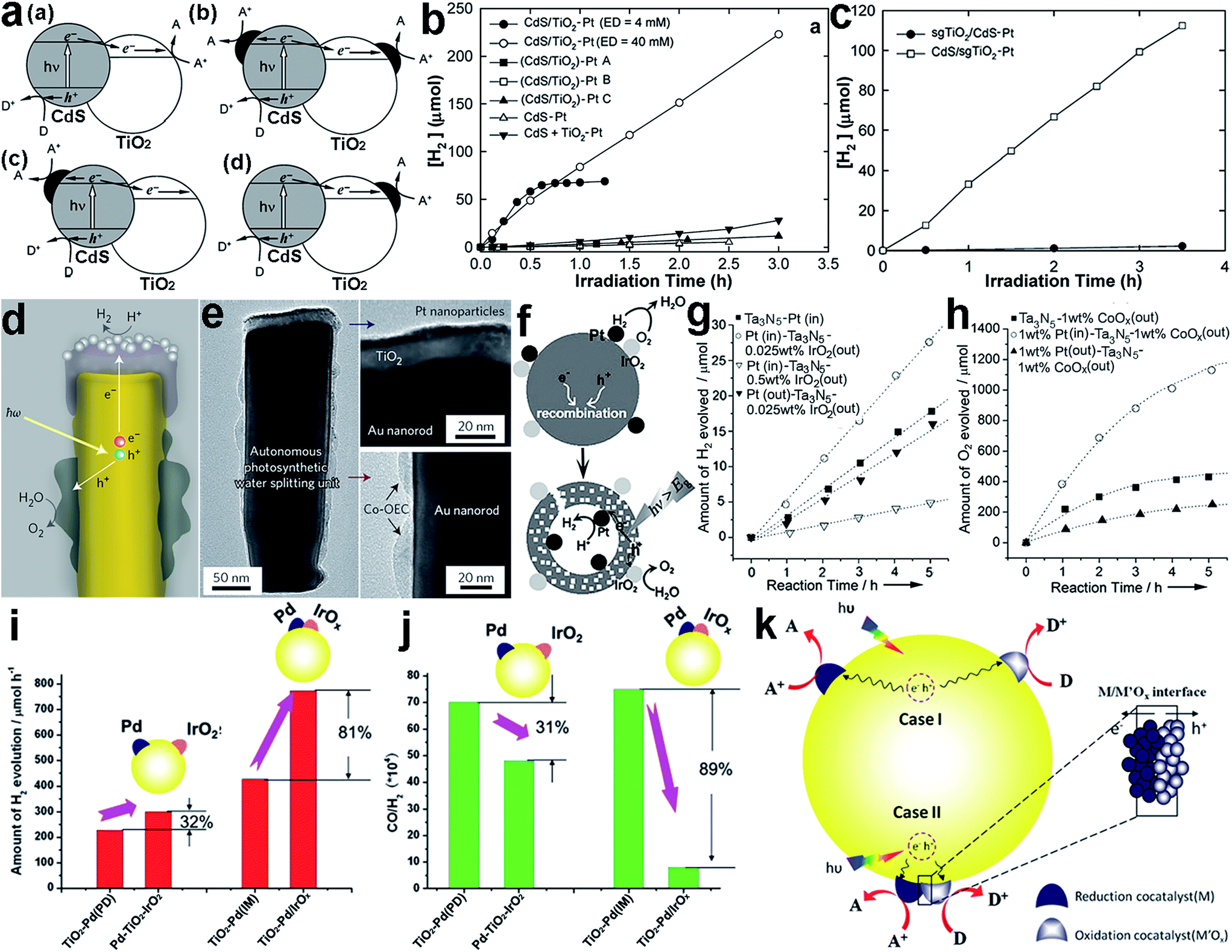 | ||
| Fig. 8 (a) Illustrative diagrams for the charge kinetics in hybrid photocatalysts: (a) CdS/TiO2, (b) (CdS/TiO2)–Pt, (c) TiO2/CdS–Pt, and (d) CdS/TiO2–Pt; (b) hydrogen production using (CdS/TiO2)–Pt and CdS/TiO2–Pt under visible light; (c) hydrogen production using sgTiO2/CdS–Pt and CdS/sgTiO2–Pt under visible light (adapted with permission from ref. 92, Copyright 2008 Royal Society of Chemistry); (d) schematic diagram of a cross-section of an individual photosynthetic unit, and (e) corresponding TEM images of Pt and Co-OEC cocatalysts in the unit (adapted with permission from ref. 93, Copyright 2013 Nature Publishing Group); (f) schematic illustration of the use of two separated cocatalysts on a core/shell Ta3N5 photocatalyst as effective charge collectors for water splitting; (g and h) time course of (g) H2 and (h) O2 evolution on core/shell Ta3N5 photocatalysts with and without spatially separated cocatalysts (adapted with permission from ref. 94, Copyright 2013 Wiley-VCH); (i and j) the difference in photocatalytic performance between Pd–TiO2–IrOx and TiO2–Pd/IrOx in (i) H2 production and (j) CO/H2 production; and (k) schematic description of the proposed mechanisms of the photocatalytic reactions on TiO2 with dual cocatalysts: (case I) spatially separated and (case II) with intimate contact (adapted with permission from ref. 95, Copyright 2014 Royal Society of Chemistry). | ||
When dual cocatalysts are involved in a photocatalytic system for reduction and oxidation reactions, the relative locations of the cocatalysts can also affect the photocatalytic performance. The spatial separation of a reduction cocatalyst from an oxidation one can not only reduce charge recombination owing to the different transfer directions of electrons and holes, but also prevent the back reaction between products given their long-distance separation. As just mentioned, the key to this system is to deposit the corresponding cocatalysts at appropriate locations on the semiconductor where electrons or holes are accumulated. Mubeen et al. designed and fabricated an autonomous plasmonic solar water splitter based on Au nanorod arrays. In the system, the TiO2 at the tips of the Au nanorods was decorated with Pt nanoparticles, as a H2 evolution cocatalyst, while Co-OEC material, as an O2 evolution cocatalyst, was deposited on the lower portion of the nanorods (Fig. 8d and e).93 The different interfacial locations of the Pt and Co-OEC cocatalysts along the Au nanorods effectively lowered the possibility of recombining plasmonic hot electrons with the remaining positive charges (i.e., hot holes) on the nanorods.
Apparently nanorods provide an anisotropic platform for separating electrons from holes, designating the locations for reduction and oxidation cocatalysts. However, the anisotropy of semiconductor nanostructures is not indispensable for cocatalyst separation. For example, Wang et al. reported a SiO2/Ta3N5 core–shell photocatalyst with reduction cocatalyst Pt nanoparticles loaded on a Ta3N5 inner shell surface, and with an oxidation cocatalyst, IrO2 or CoOx, on the outer shell surface (i.e., Pt(in)–Ta3N5–MOx(out), M = Ir or Co) (Fig. 8f).94 The separated locations of the cocatalysts facilitated the migration of photoexcited electrons and holes toward the inner and outer surfaces, respectively. This design not only reduced charge recombination, but also prevented the back reaction between the newly produced H2 and O2 to form H2O. As a result, Pt(in)–Ta3N5–IrOx(out) exhibited higher H2 evolution rates than a SiO2/Ta3N5 core–shell structure with both Pt and IrOx on the outer surface (Pt(out)–Ta3N5–IrOx(out)) (Fig. 8g). Similarly, the separated loading of the Pt and CoOx cocatalysts on the inner and outer surfaces of SiO2/Ta3N5, respectively, could also improve the O2 evolution rate (Fig. 8h).
Recently, new insight into the interfacial locations of cocatalysts has been proposed by Ma et al.95 They found that intimate contact between the reduction (Pd) and oxidation (IrOx) cocatalysts on TiO2 (i.e., TiO2–Pd/IrOx) could surprisingly lead to a significant enhancement in the photocatalytic activity for H2 production via methanol reforming compared with separated Pd and IrO2 cocatalysts on TiO2 (i.e., Pd–TiO2–IrO2). As shown in Fig. 8i, with Pd–TiO2–IrO2 with separated cocatalysts the H2 production can be increased by 32% with respect to TiO2 with a single Pd cocatalyst deposited with the same photodeposition method (i.e., TiO2–Pd(PD)). In comparison, the TiO2–Pd/IrOx achieved an 81% enhancement in H2 production in reference to TiO2–Pd(IM) prepared with the same impregnation method. In addition, the Pd–IrOx/TiO2 design also achieved improved performance toward the CO/H2 ratio (Fig. 8j). They proposed that the electrons and holes could be readily separated at the interface between IrOx and Pd in this special case. Moreover, the photogenerated charges in the surface skin region would have a shorter transportation length to the surface where the reduction and oxidation cocatalysts were closely located. On the other hand, differently from water splitting, the backward reaction with the methanol reforming products (CO2 + H2) could hardly take place when the cocatalysts were in intimate contact. A similar design has also been reported for other cocatalysts. For instance, the NiOx cocatalyst has been widely used for water splitting, and most recently, it was found that this material was actually composed of Ni and NiO which could play the role of H2 and O2 evolution cocatalyst, respectively.96,97
4.3 Interfacial facets
The efficiency of charge transfer is also dependent on the facets of components used in forming the interface. First of all, the interfacial facets, which are characterized with atomic arrangements, determine the bonding situation and lattice consistency between components as well as the coupling of their electronic structures. Secondly, the energy bands of material surfaces have a strong correlation with their surface facets, resulting in a certain alignment on the interface between the electronic band structures. Taken together, the charge transfer process at the interface largely relies on the component facets. Intuitively, the interfacial structures can be tailored either through adjusting the facet of the cocatalyst or by controlling the facet of the semiconductor for the deposition of the cocatalyst.For instance, Pt nanoparticles with different exposed facets were loaded on graphene (rGO) nanosheets to form rGO–Pt{100} and rGO–Pt{111} cocatalysts, respectively.58 With Eosin Y (EY) as a photosensitizer and rGO as a conductive component, the photoinduced electrons were transferred to the Pt cocatalyst through the rGO–Pt interface for H2 evolution. Enabled through different Pt facets at the interface, the H2 evolution activity using rGO–Pt{100} was substantially higher than that with rGO–Pt{111} (Fig. 9a). With the same tunable facets, however, bare Pt{111} as a cocatalyst exhibited a higher H2 evolution activity than bare Pt{100}, manifesting the higher catalytic activity of Pt{111}. The performance of Pt{100} can be boosted via integration with rGO, because it has stronger interactions with rGO than Pt{111} enabling faster interfacial charge transfer (Fig. 9b).
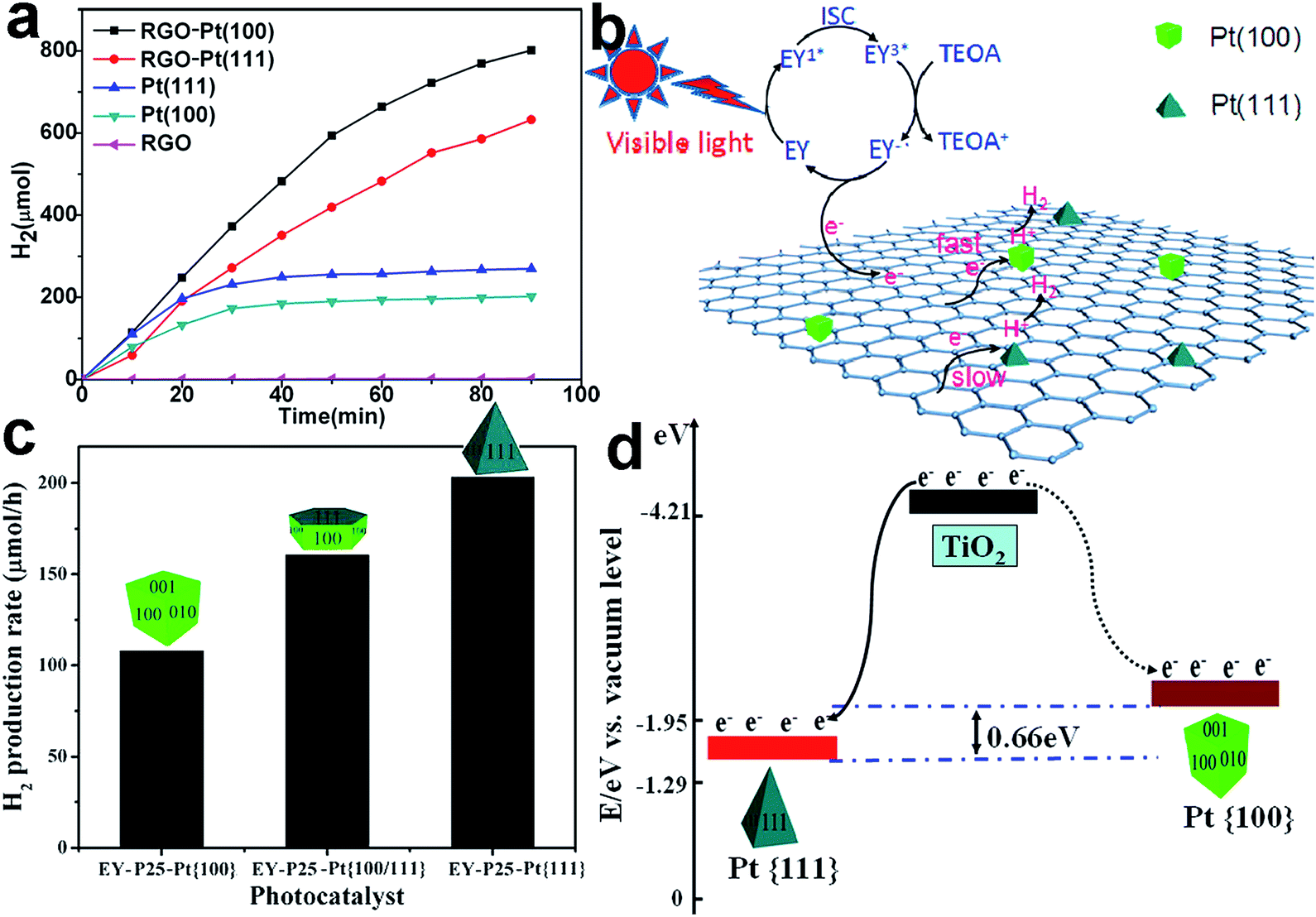 | ||
| Fig. 9 (a) H2 evolution from EY-photosensitized systems catalyzed using RGO, Pt{100}, Pt{111}, RGO–Pt{100}, and RGO–Pt{111} as cocatalysts; (b) the proposed photocatalytic mechanism for hydrogen evolution over RGO–Pt{100} and RGO–Pt{111} cocatalysts under visible-light irradiation (adapted with permission from ref. 58, Copyright 2015 American Chemical Society); (c) photocatalytic H2 evolution rates from a TEOA aqueous solution on EY–TiO2–Pt{100}, EY–TiO2–Pt{100/111}, and EY–TiO2–Pt{111} under visible-light irradiation; and (d) schematic diagram of the different energy levels in the Pt{100} (dotted curve) and Pt{111} facets (solid curve) (adapted with permission from ref. 98, Copyright 2013 American Chemical Society). | ||
In addition to the interfacial coupling, facet control also allows the tuning of the interfacial band alignments. For instance, the EY-sensitized TiO2–Pt samples with different exposed Pt facets including {100}, {100/111} and {111} were employed as photocatalysts toward H2 evolution.98 EY–TiO2–Pt{111} presented the highest H2 generation rate among the EY–TiO2–Pt samples (Fig. 9c), owing to its unique energy band alignment. The Fermi level of Pt{111} is lower than that of Pt{100}, so as to enlarge the difference between the Fermi level and the conduction band minimum (CBM) of TiO2. As a result, Pt{111} should be capable of trapping electrons from the CB of TiO2 more effectively (Fig. 9d).
It is worth noting that the variation in cocatalyst facets not only alters the interfacial structure, but also designates different exposed facets for surface reactions. This feature makes it difficult to analyze the contribution of interfacial facet design to facet-dependent photocatalytic performance. To exclude the effect from surface facets, a more straightforward method has been developed to tune interfacial facets, in which cocatalysts are selectively deposited on the different facets of a semiconductor. When semiconductor crystals are enclosed with more than one type of facet, the varied energy band structures of the surface facets may lead to spatial charge separation, so as to accumulate electrons and holes on different facets.8 In this case, the selective deposition of a reduction cocatalyst on the facet which has accumulated electrons and/or an oxidation cocatalyst on the one with holes can readily realize highly efficient interfacial charge transfer. This further promotes spatial charge separation between the facets.
For instance, Li et al. reported the selective photodeposition of metals (e.g., Pt) as reduction cocatalysts on the {010} facets of BiVO4 crystals, and metal oxides (e.g., MnOx) as oxidation cocatalysts on the {110} facets, respectively (Fig. 10a).99,100 This selective photodeposition is indeed enabled by spatial charge separation accumulating photogenerated electrons and holes on the {010} and {110} facets, respectively (Fig. 10b). In the photocatalytic water oxidation reaction, the Pt-{010}BiVO4{110}-MnOx structure exhibited remarkably higher activity in comparison with bare BiVO4 as well as BiVO4{010}–Pt and BiVO4{110}–MnOx with a single cocatalyst, manifesting the synergistic function of dual-cocatalysts (Fig. 10c). Furthermore, this study demonstrates well that interfacial facets are critical in the efficacy of cocatalysts, as a random distribution of Pt and MnOx on BiVO4 cannot offer comparable efficiency for photocatalysis.
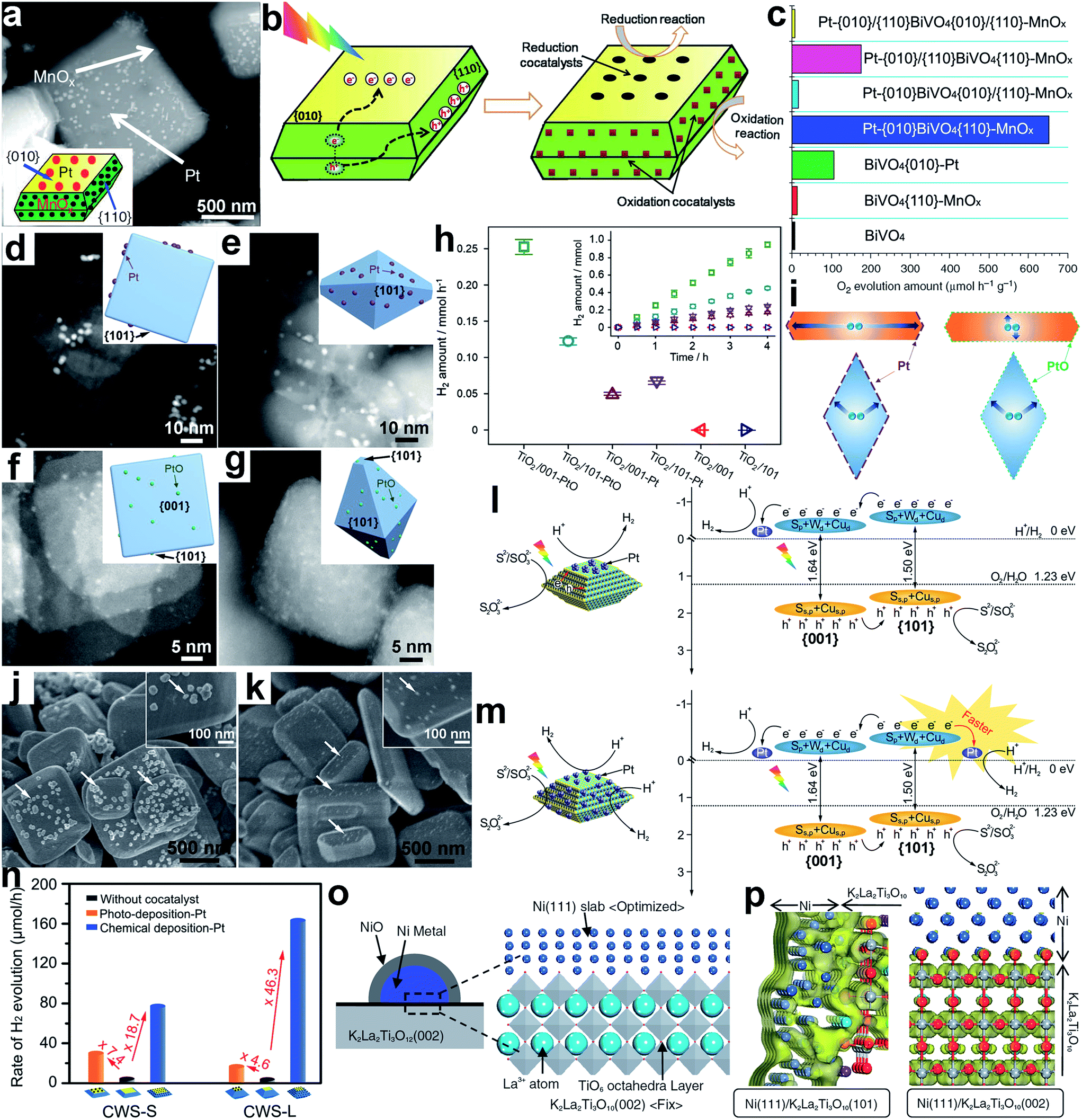 | ||
| Fig. 10 (a) SEM image and geometric models of Pt-{010}BiVO4{110}-MnOx; (b) schematic diagram illustrating the selective deposition of reduction and oxidation cocatalysts on the {010} and {110} facets of BiVO4 based on the charge separation between different facets; (c) the photocatalytic water oxidation performance of BiVO4-based photocatalysts (adapted with permission from ref. 99 and 100, Copyright 2013 Nature Publishing Group and 2014 Royal Society of Chemistry); (d–g) STEM images and geometric models of the following samples: (d) TiO2/001-Pt, (e) TiO2/101-Pt, (f) TiO2/001-PtO and (g) TiO2/101-PtO; (h) H2 evolution rates for bare TiO2 and photocatalysts loaded with PtO clusters and metallic Pt co-catalysts, respectively; (i) schematic diagram illustrating the migration of photogenerated electrons in photocatalysts (adapted with permission from ref. 104, Copyright 2015 Elsevier); (j and k) SEM images of Pt loaded CWS-S photocatalysts through (j) photodeposition and (k) chemical deposition methods; (l and m) charge kinetics of (l) CWS{001}–Pt and (m) CWS{001}/{101}-Pt photocatalysts for H2 evolution; (n) rates of photocatalytic hydrogen production over Pt deposited CWS photocatalysts (adapted with permission from ref. 105, Copyright 2015 Wiley-VCH); (o) Ni{111}/K2La2Ti3O10{002} surface unit cell model; and (p) electron density contour maps for the bottom of the CB of K2La2Ti3O10 at the Ni{111}–K2La2Ti3O10{101} and Ni{111}–K2La2Ti3O10{002} interfaces (adapted with permission from ref. 107, Copyright 2007 The Chemical Society of Japan). | ||
Nowadays, many photocatalysts have been reported through photodepositing suitable reduction (e.g., Au) and/or oxidation (e.g., Co3O4 and MnOx) cocatalysts on electron- and/or hole-accumulating facets, respectively.101,102 In addition to the spatial charge separation between different facets, photogenerated electrons and holes may also diffuse in opposite directions through spontaneous polarization, driven by an built-in dipole field in semiconductor crystals with polar facets or by a ferroelectric field in ferroelectric oxide semiconductors. This mechanism can accumulate electrons and holes at the two ends of the facets perpendicular to the field direction.8 This case calls for the necessity of selectively depositing an appropriate cocatalyst on a specific location of a facet. For instance, the asymmetric selective photodeposition of Pt and MnOx cocatalysts on the positively and negatively charged {001} facets of PbTiO3, a ferroelectric oxide semiconductor, has been demonstrated by Zhen et al.103 The resulting product showed greatly improved photocatalytic activity over those with random deposition.
When a semiconductor nanocrystal is enclosed with different facets, the traveling length of charge carriers toward the facets may depend on the nanocrystal geometry. In this case, the selective deposition of cocatalysts on the facet with a short charge transfer length may reduce electron–hole recombination and enhance the photocatalytic performance. For instance, Pt and PtO as reduction cocatalysts were dispersed on TiO2 nanosheets and octahedra dominated with {001} and {101} facets, respectively (namely, TiO2/001 and TiO2/101).104 The synthesis indicated that the metallic Pt cocatalyst was more favorably deposited on the {101} facet, while the PtO could be stabilized on both the {001} and {101} facets (Fig. 10d–g). For photocatalytic H2 evolution, TiO2/101-Pt exhibited higher activity than TiO2/001-Pt, and the activity of TiO2/001-PtO exceeded that of TiO2/101-PtO (Fig. 10h). This performance difference may originate from the reduction of charge recombination from short traveling lengths. The average electron migration length (i.e., the distance from the center of the photocatalyst to the co-catalyst) was measured to be 2 nm in TiO2/001-PtO, 25 nm in TiO2/001-Pt, and 12 nm in both TiO2/101-PtO and TiO2/101-Pt. As a result, the TiO2/101-Pt and TiO2/001-PtO greatly shortened the migration path of photogenerated electrons as compared to TiO2/001-Pt and TiO2/101-PtO (Fig. 10i), reducing the probability of charge recombination.
As the energy band structure of the semiconductor surface depends on the facets, face control would also alter interfacial band alignments, altering the charge transfer efficiency at the interface. In a typical example, for Cu2WS4 (CWS) decahedra, the CBM and valance band maximum (VBM) of the {101} facets are 80 meV and 60 meV higher than those of the {001} facets, respectively, thus resulting in spatial charge separation between the facets.105,106 Using a photodeposition method, a Pt cocatalyst was preferentially formed on the {001} facets owing to the accumulation of photogenerated electrons; the photo-free chemical deposition resulted in Pt nucleation at both the {001} and {101} facets (Fig. 10j and k).105 As illustrated in Fig. 10l, the spatial charge separation in CWS{001}–Pt drove the photogenerated electrons to move toward the Pt cocatalyst for H+ reduction, while the holes transferred toward the uncovered {101} facets to oxidize the Na2S/Na2SO3 sacrificial reagent. In comparison, CWS{001}/{101}-Pt contained one additional CWS{101}–Pt interface (Fig. 10m). As the {101} facet was rich with holes, it is not expected to be an ideal facet for loading Pt cocatalysts. In the practical photocatalytic measurements, however, CWS{001}/{101}-Pt exhibited notably higher H2 evolution activity than CWS{001}–Pt (Fig. 10n), and this activity was further promoted by increasing the {101}/{001} ratio from CWS-S to CWS-L. This indicates that the catalytic activity of Pt cocatalysts on the {101} facet should be higher than on the {001} facet (Fig. 10n). As revealed from energy band analysis, the elevated CB level of the CWS{101} facet in comparison with the {001} facet offered a larger potential difference, so that the excited electrons would more easily jump from the CWS{101} facets to the Pt cocatalysts. This feature enabled faster interfacial electron transfer toward improved photocatalysis (Fig. 10m).
Similarly to cocatalysts, altering the facets of semiconductors also brings about variations in interfacial coupling with the cocatalyst, which will influence the charge transfer efficiency across the interface. For instance, a Ni@NiO core–shell cocatalyst could be selectively loaded on the {002} and {101} facets of K2La2Ti3O10 photocatalysts for water splitting.107 The interfacial electronic structures between Ni{111} and the different facets of K2La2Ti3O10 were investigated using two-dimensional surface model density functional theory (DFT) calculations (Fig. 10o). As indicated from the electron density contour maps for K2La2Ti3O10 CBM (Fig. 10p), the Ni3d + Ti3d hybrid orbitals spread from the interface region to the Ni bulk region in the K2La2Ti3O10{101}–Ni{111} interface, whereas the K2La2Ti3O10{002}–Ni{111} interface localizes electron density within K2La2Ti3O10. This suggests that electron transfer at the K2La2Ti3O10{101}–Ni{111} interface can work more smoothly.
5. Simultaneous control over the cocatalyst surface and interface for photocatalysis
In Sections 3. and 4., surface and interface design in cocatalysts have been discussed, respectively. As briefly mentioned above, interfacial charge transfer and surface activation reactions have substantial interplay and thus are entangled together, which highlights the necessity of simultaneously controlling surface and interface parameters. If achieved, this design will allow the improvement of both charge trapping and surface activation in a single system, which would be a promising approach for high-performance photocatalysts. Certainly the parameters to be controlled in the co-design of surface and interface are quite similar to those mentioned in Sections 3. and 4. However, given the entangled relationships between surface and interface parameters, we have to more systematically consider their interplay as well as find out new approaches. For instance, it was reported that, in comparison with Pt{100}, Pt{111} cannot only trap electrons from TiO2 more effectively, but also provide more reaction sites for water reduction.98 In practical application, however, the corresponding design has not shown its strong advantages, as a high-quality interface may not be formed based on the selected cocatalyst surface. For this reason, it is imperative to develop new methods, theories and mechanisms to circumvent undesired situations.Recently, Su et al. realized the co-design of surface and interface by optimizing the electronic properties of metal cocatalysts through compositional and structural fine-tuning.108 In a semiconductor (TiO2)–metal cocatalyst system, the interfacial charge transfer from the metal to the electron acceptor (A) is a very slow process as compared to from TiO2 to the metal. For this reason, the electrons accumulated in the metal may get trapped via reverse transportation to the trap states (et) of the semiconductor (Fig. 11a). Thus a fast reduction reaction (high kred) on the surface and a slow reverse transfer process (low krev) for the trapped photogenerated electrons at the interface are both required for optimal performance. Au nanoparticles that were deposited on TiO2 could offer a large kred and krev, whereas Pd on TiO2 showed the opposite. To optimize both the kred and krev, Au- and Pd-based nanoparticles in a range of specific random alloy and core–shell configurations, such as Au1Pd1 random alloy (Fig. 11b), Aushell–Pdcore (AusPdc, Fig. 11c) and Pdshell–Aucore (PdsAuc, Fig. 11d) nanoparticles, were formed and supported on TiO2 as cocatalysts. By probing the kinetics under constant irradiation, it was revealed that an increase in Pd content in the alloy nanoparticles resulted in a ∼33–50% reduction in kred as well as an exponential decrease in krev (Fig. 11e). Conversely, PdsAuc maintained a relatively high kred but a very low krev relative to AusPdc. As a result, the TiO2–PdsAuc exhibited higher H2 evolution rates in comparison with the TiO2–AuPd alloy and TiO2–AusPdc (Fig. 11f).
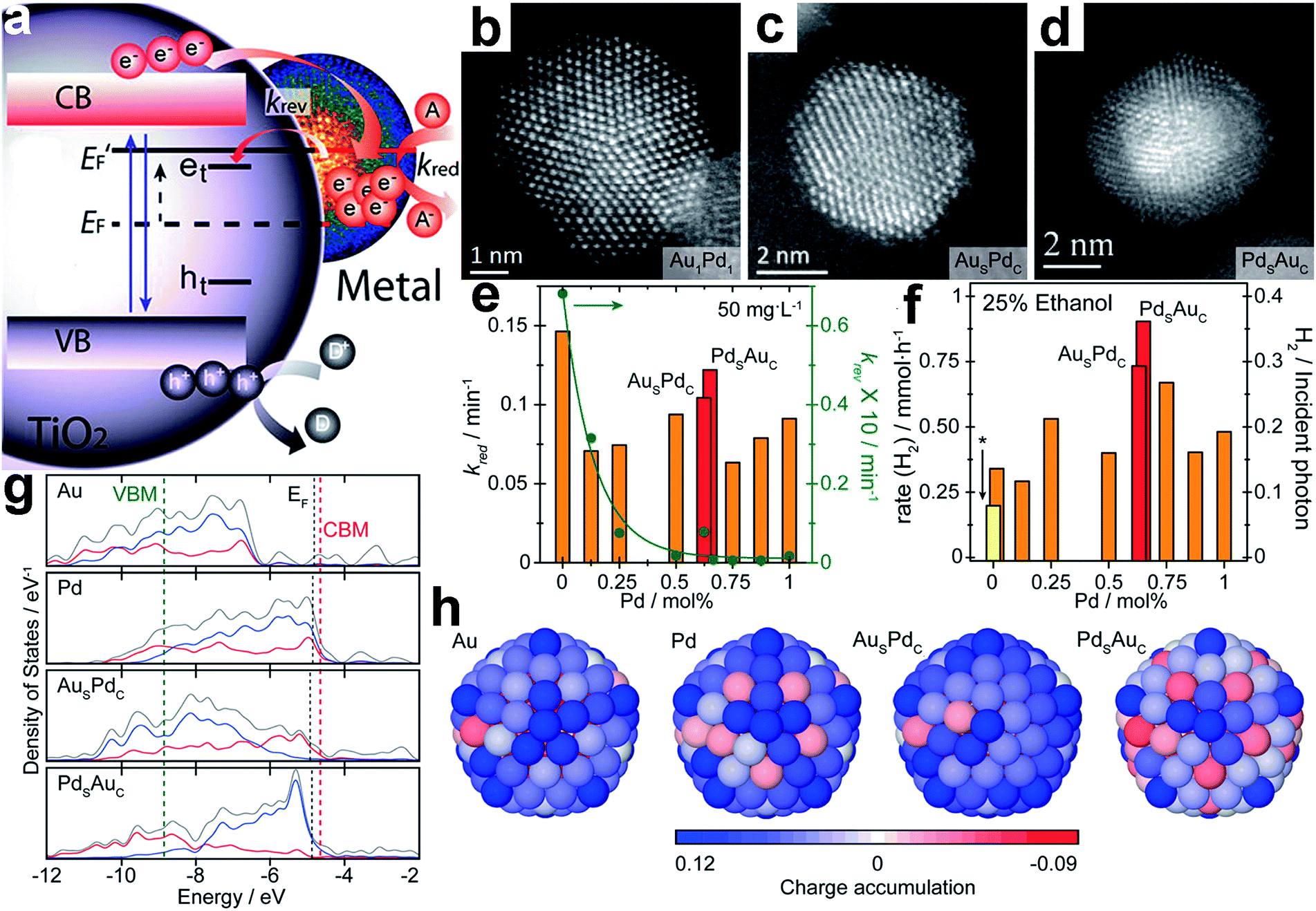 | ||
| Fig. 11 (a) Schematic diagram of energy levels in a semiconductor (TiO2)–metal cocatalyst system under UV irradiation: EF and E′F represent the Fermi level of the system before and after irradiation; the trap states of excited electrons and holes are indicated as et and ht; kred and krev denote the reduction rate, and the reversed trapping rate, respectively; (b–d) representative HAADF-STEM images of (b) a Au1Pd1 random alloy particle, (c) a Aushell–Pdcore particle, and (d) a Pdshell–Aucore particle; (e) rate constants kred and krev as a function of Pd concentration under constant irradiation; (f) photocatalytic H2 production from a 25 vol% ethanol solution using random alloys and core–shell Au–Pd cocatalysts supported on TiO2; (g) electronic density of states (DOS) profiles of 147-atom icosahedral clusters of Au, Pd, AusPdc, and PdsAuc: the total valence DOS (gray) is complemented by contributions from the d-orbitals in both the core (red) and outer shell (blue) region; the EF of the metal clusters, and the CBM and VBM of TiO2 are indicated with black, red and green dashed lines, respectively; (h) charge localization from the view of the {100} surface of 147-atom icosahedral clusters of Au, Pd, AusPdc, and PdsAuc: the blue and red colors represent atoms with electron accumulation and depletion, respectively (adapted with permission from ref. 108, Copyright 2014 American Chemical Society). | ||
Fig. 11g shows the electronic density of states (DOS) profiles of 147-atom icosahedral clusters of Au, Pd, AusPdc, and PdsAuc, respectively. It depicts that Pd clusters possess more unoccupied states just above the Fermi level (EF) than Au, favorable for electron trapping. In the case of the core–shell structure, the number of unoccupied surface states just above EF increases with the Pd concentration on the surface. Compared to <8% for AusPdc, PdsAuc shows that >60% of all states just above EF are surface-localized d-orbitals, which are highly beneficial for extending the lifetime of the photoexcited states transferred from the CB of TiO2. Furthermore, charge localization on the surface of the icosahedral clusters shows that net electron accumulation predominantly occurs throughout the shell, whereas for PdsAuc, a net accumulation of charge takes place primarily on the vertex sites (Fig. 11h). On the other hand, charge depletion is prominent for the core regions of all the cocatalysts except PdsAuc owing to the electronegative and capacitive properties of the Au core. It was proposed that the reduced surface accumulation of electrons in PdsAuc facilitated the storage and release of photoexcited electrons toward surface catalysis.
In another case, we achieved the co-design of a cocatalyst surface and interface by employing a different mechanism – interfacial charge polarization. To implement the mechanism, atomically controlled Pd@Pt core–shell cocatalysts were fabricated on TiO2 nanosheets.65 Specifically, we used Pd nanocubes supported on TiO2 nanosheets (TiO2–Pd) as precursors, and Pt shells with a controllable thickness could be selectively coated on the Pd cubes to form TiO2–Pd@Pt photocatalysts. As shown in Fig. 12a and b, TiO2–Pd@Pt photocatalysts with three (TiO2–Pd@Pt3L) and ten (TiO2–Pd@Pt10L) Pt atomic layers on average were synthesized by altering the TiO2–Pd/Pt precursor ratios. As such, the surface of the cocatalysts changed from Pd{100} to Pt{100} with higher H2 evolution activity. Furthermore, the Pt nanocubes with the same Pt{100} exposed were also loaded on TiO2 nanosheets to provide a reference sample (TiO2–Pt). From the photocurrent versus time (I–t) curves in Fig. 12c, the photocurrents turn out to be in the order of TiO2–Pd < TiO2–Pt < TiO2–Pd@Pt10L < TiO2–Pd@Pt3L, suggesting more efficient electron–hole separation by the Pd@Pt cocatalyst. The same order was also recognized for hydrogen production rates, but the improvement in hydrogen production from the cocatalyst design appears to be more significant than the photocurrent (Fig. 12d).
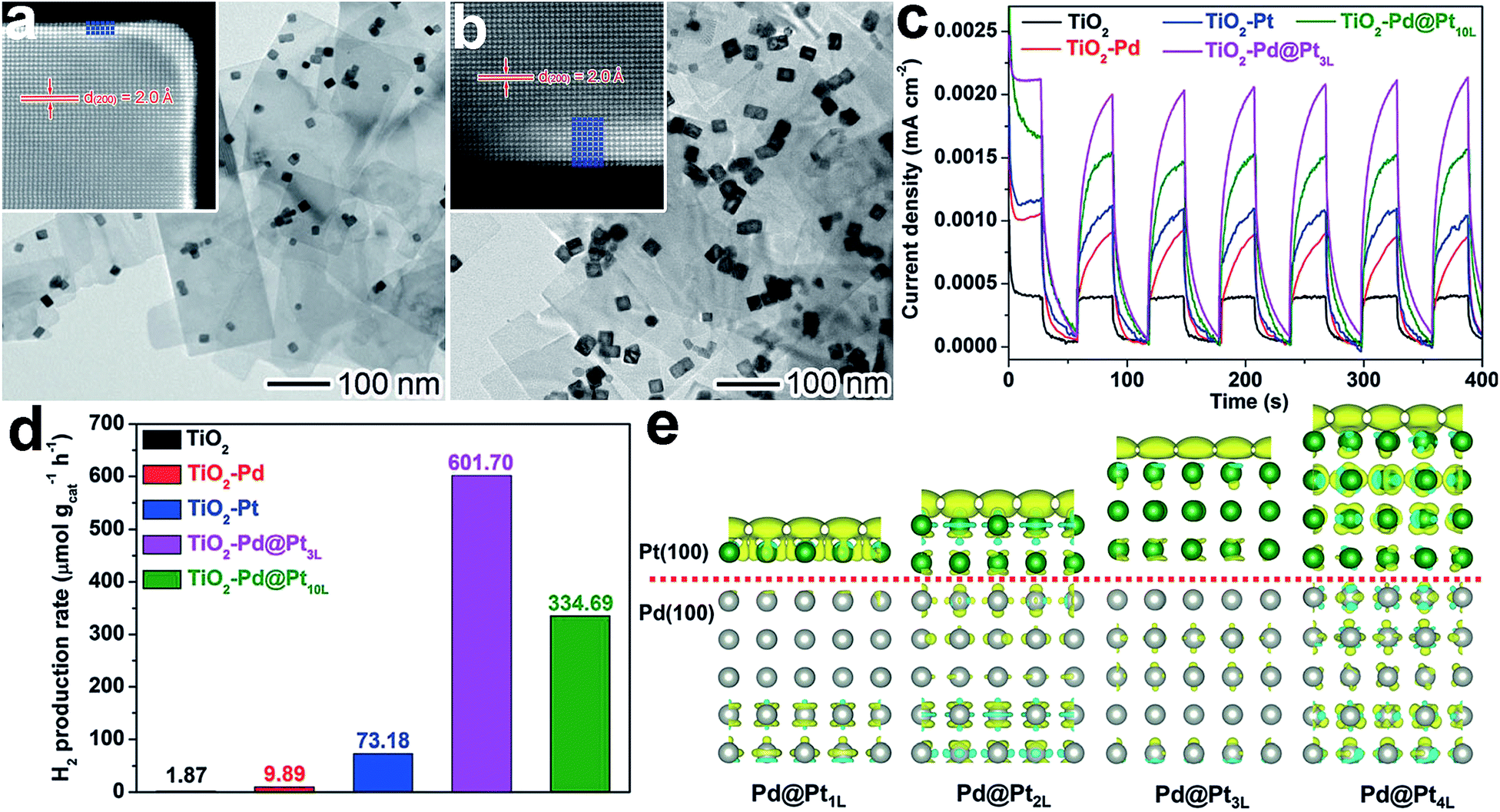 | ||
| Fig. 12 (a and b) TEM images and atomic-resolution HAADF-STEM images of (a) Pd@Pt3L and (b) Pd@Pt10L quasi-core–shell cocatalysts supported on TiO2 nanosheets; (c and d) I–t curves (c) and photocatalytic H2 evolution (d) for TiO2–Pd@Pt3L and TiO2–Pd@Pt10L in comparison with bare TiO2 nanosheets, TiO2–Pd, and TiO2–Pt hybrid structures under UV light irradiation; (e) differential charge density determined using first-principles simulations for Pt{100} shells on Pd{100} substrates in the presence of one additional electron: the olive and cyan colors represent an increase and decrease in electron density, respectively (adapted with permission from ref. 65, Copyright 2015 Wiley-VCH). | ||
The enhancement of photocatalytic performance with the Pd@Pt cocatalyst resulted from charge polarization at the Pd–Pt interface. Owing to the different work functions of Pd and Pt, electron migration across the interface from Pd to Pt will equilibrate their electron Fermi distributions. When TiO2 was photoexcited, interfacial charge transfer would occur from TiO2 to Pd driven by a TiO2–Pd Schottky junction. Meanwhile, the interfacial polarization would serve as a new driving force for the migration of photogenerated electrons from the Pd to Pt surface, improving the charge separation in the entire system. Furthermore, the interfacial polarization also led to the accumulation of electrons on the Pt surface. Both the electron accumulation and lattice strain at the Pd–Pt interface facilitated H2O adsorption. As a result, the charge polarization does not only improve the charge separation, but it also enhances the H2O adsorption. It should be noted that the interfacial polarization effect decays with an increase in Pt shell thickness (Fig. 12e). For this reason, the shell thickness has to be kept within a few atomic layers. This delicate design enables a boosting of the photocatalytic performance and a reduction in the amount of expensive Pt used.
6. Summary and outlook
The surface and interface design of cocatalysts would be a promising route to fabricate high-performance photocatalysts through maximizing the efficacy of cocatalysts. In this review, we have highlighted the emerging horizons of cocatalyst design based on surface and interface adjustment. On one hand, surface parameters such as the composition, facets, phases and defects have been tailored to enhance catalytic reactions on cocatalyst surfaces. On the other hand, interface parameters including interfacial composition, location and facets have been optimized to improve the charge transfer across the interface of cocatalysts with semiconductors. With novel methods, theories, and mechanisms (e.g., the interfacial polarization effect) implemented in cocatalyst design, surface and interface control has enabled the enhancement of surface reactions and interfacial charge transfer simultaneously. This set of designs not only deepens our understanding of fundamental aspects, but also provides technical approaches to significantly enhance the activity and selectivity in water splitting and CO2 reduction.Although great achievement and remarkable progress have been made in the surface and interface design of cocatalysts, there is a long way to go toward the practical use of photocatalysis in industry and our lives. Firstly, most of the previous reports on cocatalyst design were to tailor only one surface or interface parameter. It remains a grand challenge to design cocatalysts taking more parameters into account. The bottlenecks include the interplaying effects of the parameters and the limited synthetic methods for realizing complicated designs. Secondly, some experimental results achieved in the designs still remain elusive. In many cases, existing observations seem contradictory between different reports. For instance, it has been reported that spatially separating reduction and oxidation cocatalysts and having them in intimate contact can both enhance charge separation;94,95 and that surface reactions on reduction cocatalysts can be facilitated by both reducing electron density or accumulating electrons on the surface.65,108 Thirdly, advanced characterization techniques are lacking to monitor the processes of surface reactions and interfacial charge transfer at high spatial and temporal resolution. Dynamic evolution at electron and molecular levels will provide important information for establishing the relationship between surface/interface parameters and catalytic performance. The above bottlenecks call for research at the intersection of precisely controlled synthesis, theoretical simulations and advanced spectroscopic characterization.8 Multidisciplinary collaborations at the intersection of these will offer the research community the capabilities to tailor cocatalyst structures with atomic precision and to understand the underlying mechanisms at the electron and molecular level. As soon as these existing bottlenecks are overcome, the more rational and systematic design of cocatalyst surfaces and interfaces would serve as a strong driving force toward the fabrication of highly efficient photocatalysts for solar fuel production.
Acknowledgements
This work was financially supported by the 973 Program (No. 2014CB848900), NSFC (No. 21471141, U1532135), the Recruitment Program of Global Experts, the CAS Hundred Talent Program, Hefei Science Center CAS (No. 2015HSC-UP009, 2015HSC-UP020), Fundamental Research Funds for the Central Universities (No. WK2060190025, WK2310000035), Zhejiang Provincial Natural Science Foundation (No. LQ16B010001), and the Open Research Fund of Top Key Discipline of Chemistry in Zhejiang Provincial Colleges and Key Laboratory of the Ministry of Education for Advanced Catalysis Materials (Zhejiang Normal University) (ZJHX201507).Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS PubMed.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- X. Chen, S. Chen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503 CrossRef CAS PubMed.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372 CrossRef CAS PubMed.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900 CrossRef CAS PubMed.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787 RSC.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893 RSC.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987 CrossRef CAS PubMed.
- L. Wang, F. Dionigi, N. T. Nguyen, R. Kirchgeorg, M. Gliech, S. Grigorescu, P. Strasser and P. Schmuki, Chem. Mater., 2015, 27, 2360 CrossRef CAS.
- S. Bai and Y. Xiong, Chem. Commun., 2015, 51, 10261 RSC.
- S. Bai, W. Jiang, Z. Li and Y. Xiong, ChemNanoMat, 2015, 1, 223 CrossRef CAS.
- Q. Zhai, S. Xie, W. Fan, Q. Zhang, Y. Wang, W. Deng and Y. Wang, Angew. Chem., Int. Ed., 2013, 52, 5776 CrossRef CAS PubMed.
- S. Bai, X. Wang, C. Hu, M. Xie, J. Jiang and Y. Xiong, Chem. Commun., 2014, 50, 6094 RSC.
- S. Chen, S. Shen, G. Liu, Y. Qi, F. Zhang and C. Li, Angew. Chem., Int. Ed., 2015, 54, 3047 CrossRef CAS PubMed.
- M. Guan, C. Xiao, J. Zhang, S. Fan, R. An, Q. Cheng, J. Xie, M. Zhou, B. Ye and Y. Xie, J. Am. Chem. Soc., 2013, 135, 10411 CrossRef CAS PubMed.
- R. Long, S. Zhou, B. J. Wiley and Y. Xiong, Chem. Soc. Rev., 2014, 43, 6288 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- M. Yoshida, A. Yamakata, K. Takanabe, J. Kubota, M. Osawa and K. Domen, J. Am. Chem. Soc., 2009, 131, 13218 CrossRef CAS PubMed.
- F. N. Sayed, O. D. Jayakumar, R. Sasikala, R. M. Kadam, S. R. Bharadwaj, L. Kienle, U. Schurmann, S. Kaps, R. Adelung, J. P. Mittal and A. K. Tyagi, J. Phys. Chem. C, 2012, 116, 12462 CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806 CrossRef CAS PubMed.
- C. G. Silva, R. Juarez, T. Marino, R. Molinari and H. Garcia, J. Am. Chem. Soc., 2011, 133, 595 CrossRef PubMed.
- P. D. Tran, L. Xi, S. K. Batabyal, L. H. Wong, J. Barber and J. S. C. Loo, Phys. Chem. Chem. Phys., 2012, 14, 11596 RSC.
- W. J. Foo, C. Zhang and G. W. Ho, Nanoscale, 2013, 5, 759 RSC.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrovic, D. Volbers, R. Wyrwich, M. Doblinger, A. S. Susha, A. L. Rogach, F. Jackel, J. K. Stolarczyk and J. Feldmann, Nat. Mater., 2014, 13, 1013 CrossRef CAS PubMed.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176 CrossRef CAS PubMed.
- J. Yuan, J. Wen, Y. Zhong, X. Li, Y. Fang, S. Zhang and W. Liu, J. Mater. Chem. A, 2015, 3, 18244 CAS.
- X. Zong, J. Han, G. Ma, H. Yan, G. Wu and C. Li, J. Phys. Chem. C, 2011, 115, 12202 CAS.
- Z. H. Shah, J. Wang, Y. Ge, C. Wang, W. Mao, S. Zhang and R. Lu, J. Mater. Chem. A, 2015, 3, 3568 CAS.
- C. C. Hu and H. S. Teng, J. Catal., 2010, 272, 1 CrossRef CAS.
- S. Cao, Y. Chen, C. C. Hou, X. J. Lv and W. F. Fu, J. Mater. Chem. A, 2015, 3, 6096 CAS.
- Q. Yue, Y. Wan, Z. Sun, X. J. Wu, Y. Yuan and P. Du, J. Mater. Chem. A, 2015, 3, 16941 CAS.
- Z. Sun, H. Zheng, J. Li and P. Du, Energy Environ. Sci., 2015, 8, 2668 CAS.
- G. Xie, K. Zhang, B. Guo, Q. Liu, L. Fang and J. R. Gong, Adv. Mater., 2013, 25, 3820 CrossRef CAS PubMed.
- A. Ye, W. Fan, Q. Zhang, W. Deng and Y. Wang, Catal. Sci. Technol., 2012, 2, 969 CAS.
- J. Yu, K. Wang, W. Xiao and B. Cheng, Phys. Chem. Chem. Phys., 2014, 16, 11492 RSC.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863 CrossRef CAS PubMed.
- I. H. Tseng, J. C. S. Wu and H. Y. Chou, J. Catal., 2004, 221, 432 CrossRef CAS.
- P. W. Pan and Y. W. Chen, Catal. Commun., 2007, 8, 1546 CrossRef CAS.
- S. C. Yan, S. X. Ouyang, J. Gao, M. Yang, J. Y. Feng, X. X. Fan, L. J. Wan, Z. S. Li, J. H. Ye, Y. Zhou and Z. G. Zou, Angew. Chem., Int. Ed., 2010, 49, 6400 CrossRef CAS PubMed.
- S. Wang, Z. Ding and X. Wang, Chem. Commun., 2015, 51, 1517 RSC.
- S. Wang, Y. Hou and X. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 4327 CAS.
- L. Liu, Z. Ji, W. Zou, X. Gu, Y. Deng, F. Gao, C. Tang and L. Dong, ACS Catal., 2013, 3, 2052 CrossRef CAS.
- R. Asai, H. Nemoto, Q. Jia, K. Saito, A. Iwase and A. Kudo, Chem. Commun., 2014, 50, 2543 RSC.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348 CrossRef CAS PubMed.
- B. Ma, J. Yang, H. Han, J. Wang, X. Zhang and C. Li, J. Phys. Chem. C, 2010, 114, 12818 CAS.
- R. Abe, M. Higashi and K. Domen, J. Am. Chem. Soc., 2010, 132, 11828 CrossRef CAS PubMed.
- E. M. P. Steinmiller and K. S. Choi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20633 CrossRef CAS PubMed.
- D. Wang, R. Li, J. Zhu, J. Shi, J. Han, X. Zong and C. Li, J. Phys. Chem. C, 2012, 116, 5082 CAS.
- G. Ai, R. Mo, H. Li and J. Zhong, Nanoscale, 2015, 7, 6722 RSC.
- K. Maeda, A. K. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. L. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096 CrossRef CAS PubMed.
- S. S. K. Ma, K. Maeda, R. Abe and K. Domen, Energy Environ. Sci., 2012, 5, 8390 CAS.
- A. Tanaka, S. Sakaguchi, K. Hashimoto and H. Kominami, ACS Catal., 2013, 3, 79 CrossRef CAS.
- A. Tanaka, K. Nakanishi, R. Hamada, K. Hashimoto and H. Kominami, ACS Catal., 2013, 3, 1886 CrossRef CAS.
- W. Jiang, S. Bai, L. Wang, X. Wang, L. Yang, Y. Li, D. Liu, X. Wang, Z. Li, J. Jiang and Y. Xiong, Small, 2016, 12, 1640 CrossRef CAS PubMed.
- Z. Zheng, T. Tachikawa and T. Majima, J. Am. Chem. Soc., 2014, 136, 6870 CrossRef CAS PubMed.
- W. Zhang, Y. Li, X. Zeng and S. Peng, Sci. Rep., 2015, 5, 10589 CrossRef CAS PubMed.
- Z. Li, Q. Wang, C. Kong, Y. Wu, Y. Li and G. Lu, J. Phys. Chem. C, 2015, 119, 13561 CAS.
- C. Kong, S. Min and G. Lu, Chem. Commun., 2014, 50, 9281 RSC.
- H. Du, X. Xie, Q. Zhu, L. Lin, Y. F. Jiang, Z. K. Yang, X. Zhou and A. W. Xu, Nanoscale, 2015, 7, 5752 RSC.
- Y. P. Xie, G. Liu, G. Q. Lu and H. M. Cheng, Nanoscale, 2012, 4, 1267 RSC.
- K. Maeda, M. Higashi, D. Lu, R. Abe and K. Domen, J. Am. Chem. Soc., 2010, 132, 5858 CrossRef CAS PubMed.
- S. Xie, Y. Wang, Q. Zhang, W. Deng and Y. Wang, ACS Catal., 2014, 4, 3644 CrossRef CAS.
- S. Xie, Y. Wang, Q. Zhang, W. Fan, W. Deng and Y. Wang, Chem. Commun., 2013, 49, 2451 RSC.
- S. Bai, L. Yang, C. Wang, Y. Lin, J. Lu, J. Jiang and Y. Xiong, Angew. Chem., Int. Ed., 2015, 54, 14810 CrossRef CAS PubMed.
- C. W. Tsai, H. M. Chen, R. S. Liu, K. Asakura and T. S. Chan, J. Phys. Chem. C, 2011, 115, 10180 CAS.
- K. Maeda, N. Sakamoto, T. Ikeda, H. Ohtsuka, A. Xiong, D. Lu, M. Kanehara, T. Teranishi and K. Domen, Chem.–Eur. J., 2010, 16, 7750 CrossRef CAS PubMed.
- Z. Wang, G. Liu, C. Ding, Z. Chen, F. Zhang, J. Shi and C. Li, J. Phys. Chem. C, 2015, 119, 19607 CAS.
- D. Jiang, L. Chen, J. Xie and M. Chen, Dalton Trans., 2014, 43, 4878 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575 CrossRef CAS PubMed.
- K. Chang, Z. Mei, T. Wang, Q. Kang, S. Ouyang and J. Ye, ACS Nano, 2014, 8, 7078 CrossRef CAS PubMed.
- Q. Li, C. Cui, H. Meng and J. Yu, Chem.–Asian J., 2014, 9, 1766 CrossRef CAS PubMed.
- G. W. Busser, B. Mei, P. Weide, P. C. K. Vesborg, K. Stuhrenberg, M. Bauer, X. Huang, M. G. Willinger, I. Chorkendorff, R. Schlogl and M. Muhler, ACS Catal., 2015, 5, 5530 CrossRef CAS.
- Y. H. Li, J. Xing, Z. J. Chen, Z. Li, F. Tian, L. R. Zheng, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, Nat. Commun., 2013, 4, 2500 Search PubMed.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151 CAS.
- D. Wang, A. Pierre, M. G. Kibria, K. Cui, X. Han, K. H. Bevan, H. Guo, S. Paradis, A. R. Hakima and Z. Mi, Nano Lett., 2011, 11, 2353 CrossRef CAS PubMed.
- M. G. Kibria, S. Zhao, F. A. Chowdhury, Q. Wang, H. P. T. Nguyen, M. L. Trudeau, H. Guo and Z. Mi, Nat. Commun., 2014, 5, 3825 CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655 CrossRef CAS.
- G. Zhang, G. Li and X. Wang, ChemCatChem, 2015, 7, 2864 CrossRef CAS.
- L. Zhang, Q. Liu, T. Aoki and P. A. Crozier, J. Phys. Chem. C, 2015, 119, 7207 CAS.
- Q. Liu, L. Zhang and P. A. Crozier, Appl. Catal., B, 2015, 172, 58 CrossRef.
- T. Takata, C. Pan and K. Domen, ChemElectroChem, 2016, 3, 31 CrossRef CAS.
- C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 2955 CrossRef CAS PubMed.
- T. Takata, C. Pan, M. Nakabayashi, N. Shibata and K. Domen, J. Am. Chem. Soc., 2015, 137, 9627 CrossRef CAS PubMed.
- C. Kong, S. Min and G. Lu, Chem. Commun., 2014, 50, 9281 RSC.
- S. Bai, M. Xie, Q. Kong, W. Jiang, R. Qiao, Z. Li, J. Jiang and Y. Xiong, Part. Part. Syst. Charact. DOI:1002/ppsc.201500239.
- E. Aronovitch, P. Kalisman, S. Mangel, L. Houben, L. Amirav and M. Bar-Sadan, J. Phys. Chem. Lett., 2015, 6, 3760 CrossRef CAS PubMed.
- M. Luo, W. Yao, C. Huang, Q. Wu and Q. Xu, J. Mater. Chem. A, 2015, 3, 13884 CAS.
- G. Kovacs, S. Fodor, A. Vulpoi, K. Schrantz, A. Dombi, K. Hernadi, V. Danciu, Z. Pap and L. Baia, J. Catal., 2015, 325, 156 CrossRef CAS.
- S. Bai, L. Wang, X. Chen, J. Du and Y. Xiong, Nano Res., 2015, 8, 175 CrossRef CAS.
- J. Xiong, Y. Liu, D. Wang, S. Liang, W. Wu and L. Wu, J. Mater. Chem. A, 2015, 3, 12631 CAS.
- H. Park, W. Choi and M. R. Hoffmann, J. Mater. Chem., 2008, 18, 2379 RSC.
- S. Mubeen, J. Lee, N. Singh, S. Kramer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247 CrossRef CAS PubMed.
- D. Wang, T. Hisatomi, T. Takata, C. Pan, M. Katayama, J. Kubota and K. Domen, Angew. Chem., Int. Ed., 2013, 52, 11252 CrossRef CAS PubMed.
- Y. Ma, R. Chong, F. Zhang, Q. Xu, S. Shen, H. Han and C. Li, Phys. Chem. Chem. Phys., 2014, 16, 17734 RSC.
- T. K. Townsend, N. D. Browning and F. E. Osterloh, Energy Environ. Sci., 2012, 5, 9543 CAS.
- Q. Zhang, Z. Li, S. Wang, R. Li, X. Zhang, Z. Liang, H. Han, S. Liao and C. Li, ACS Catal., 2016, 6, 2182 CrossRef CAS.
- E. Cui and G. Lu, J. Phys. Chem. C, 2013, 117, 26415 CAS.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- R. Li, H. Han, F. Zhang, D. Wang and C. Li, Energy Environ. Sci., 2014, 7, 1369 CAS.
- R. Li, X. Tao, R. Chen, F. Fan and C. Li, Chem.–Eur. J., 2015, 21, 14337 CrossRef CAS PubMed.
- L. Zhang, W. Wang, S. Sun, D. Jiang and E. Gao, Appl. Catal., B, 2015, 162, 470 CrossRef CAS.
- C. Zhen, J. C. Yu, G. Liu and H. M. Cheng, Chem. Commun., 2014, 50, 10416 RSC.
- Y. H. Li, C. Peng, S. Yang, H. F. Wang and H. G. Yang, J. Catal., 2015, 330, 120 CrossRef CAS.
- B. Wang, M. Liu, Z. H. Zhou and L. Guo, Adv. Sci., 2015, 2, 1500153 Search PubMed.
- N. Li, M. Liu, Z. Zhou, J. Zhou, Y. Sun and L. Guo, Nanoscale, 2014, 6, 9695 RSC.
- Y. Shimodaira, A. Kudo and H. Kobayashi, Chem. Lett., 2007, 36, 170 CrossRef CAS.
- R. Su, R. Tiruvalam, A. J. Logsdail, Q. He, C. A. Downing, M. T. Jensen, N. Dimitratos, L. Kesavan, P. P. Wells, R. Bechstein, H. H. Jensen, S. Wendt, C. R. A. Catlow, C. J. Kiely, G. J. Hutchings and F. Besenbacher, ACS Nano, 2014, 8, 3490 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |