
A photo- and thermo-responsive star-shaped diblock copolymer with a porphyrin core prepared via consecutive ATRPs†
Abstract
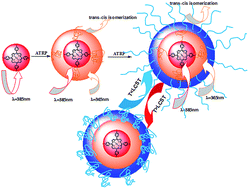
A star-shaped and diblock copolymer poly(6-[4-(4-methoxyphenylazo)phenoxy]hexylmethacrylate)s-b-poly(N-isopropylacrylamide)s with a zinc-porphyrin core (PAzo-b-PNIPAM) is synthesized via consecutive atom transfer radical polymerizations (ATRPs). The synthesis involves the preparation of the intermediate poly(6-[4-(4-methoxyphenylazo)phenoxy]hexyl methacrylate) with a zinc-porphyrin core (por-PAzo) via ATRP, and then por-PAzo is used as a macroinitiator to polymerize N-isopropylacrylamide (NIPAM) via ATRP to gain PAzo-b-PNIPAM. The structures of the target products are characterized by FT-IR, 1H NMR and UV-vis spectra, and the polydispersity index (PDI) indicates that the molecular weight distribution is narrow and the polymerization is well controlled. Furthermore, the photo- and thermo-responsive properties are also investigated in detail, which demonstrate that the star-shaped diblock copolymer PAzo-b-PNIPAM has a potential application in targeted photodynamic therapy and photo-electro informational storage.
 Please wait while we load your content...
Please wait while we load your content...

