
An ultra high performance liquid chromatography-tandem mass spectrometry method for the quantification of linagliptin in human plasma
Nagaraj Kumar Nannapaneniab,
Sunil S. Jalalpure*a,
Rajendraprasad Muppavarapub and
Sunil Kumar Sirigirib
aDr. Prabhakar Kore Basic Science Research Centre, KLE College of Pharmacy, KLE University, Nehru Nagar, Belagavi 590 010, Karnataka, India. E-mail: jalalpureesunil@rediffmail.com; Tel: +91 9448964057
bBioanalytical Research Unit, Jeevan Scientific Technology Ltd, Manikonda Jagir, Hyderabad 500 008, Telangana, India
First published on 24th June 2016
Abstract
A simple, rapid, sensitive, reliable and selective ultra high performance liquid chromatography (UHPLC)-tandem mass spectrometry (MS/MS) method was developed for the quantification of linagliptin (LGN) in human plasma. LGN and its deuterated internal standard (IS) LGN-d4 were extracted from a low-plasma sample (volume: 300 μL) by a simple liquid–liquid extraction protocol. Efficient estimation of the analyte and IS at a mean retention time (RT) of 1.75 and 1.74 min respectively, with a rapid 3.5 min run time per sample was chromatographically established on a Gemini C18 (100 mm × 4.6 mm, 3 μ) column under simple isocratic elution conditions, using a mixture of 10 mM ammonium formate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol [20:80 (v/v)] delivered at a flow rate of 0.5 mL min−1. Following the separation of the compounds, protonated precursor → product ion transitions were monitored for LGN (m/z: 473.3 → 420.1) and IS (m/z: 477.5 → 424.3) on a triple quadrupole mass spectrometer, operating in a multiple reaction monitoring (MRM) mode. The most recent regulatory guidelines were adopted during the method validation. The method demonstrated very good analyte and IS recovery (not less than 71.0%), precision (≤8.6% CV), accuracy (range: 86.7% to 95.6%) and linearity (r > 0.99) across a clinically relevant LGN plasma concentration range: 50.3 to 12115.5 pg mL−1. The validated method was successfully applied to pharmacokinetic study samples for measuring linagliptin plasma levels.
:methanol [20:80 (v/v)] delivered at a flow rate of 0.5 mL min−1. Following the separation of the compounds, protonated precursor → product ion transitions were monitored for LGN (m/z: 473.3 → 420.1) and IS (m/z: 477.5 → 424.3) on a triple quadrupole mass spectrometer, operating in a multiple reaction monitoring (MRM) mode. The most recent regulatory guidelines were adopted during the method validation. The method demonstrated very good analyte and IS recovery (not less than 71.0%), precision (≤8.6% CV), accuracy (range: 86.7% to 95.6%) and linearity (r > 0.99) across a clinically relevant LGN plasma concentration range: 50.3 to 12115.5 pg mL−1. The validated method was successfully applied to pharmacokinetic study samples for measuring linagliptin plasma levels.
1 Introduction
An astonishing figure of 415 million people are diabetic with an estimated health expenditure reaching USD 673 billion in 2015, and a dramatic increase to 642 million people could be possible by 2040, if the same trend continues. Up to 91% of affected adults have type 2 diabetes and are based in high income countries.1 DPP-4 (dipeptidyl peptidase-4) inhibitors, a relatively new class of blood glucose lowering drugs, acts by stimulating insulin secretion through the degradation of glucagon like peptide-1 (GLP-1), an endogenous substance2 and they are well tolerated as their adverse effect profile matches a placebo.3 Linagliptin (LGN) is an orally active and competitive DPP-4 enzyme inhibitor with a xanthine based chemical structure4,5 used to treat type 2 diabetics who failed to achieve glycemic control with metformin alone.6 It is approved in US, Europe, Japan and other countries7,8 and administered as a single drug as a once daily 5 mg dose in adults or in combination with metformin as a twice daily dose.9 LGN exhibits a similar pharmacokinetic (PK) profile in healthy subjects and in type 2 diabetics. Peak plasma levels (Tmax) were achieved approximately 1.5 hours after administration of a single 5 mg tablet (in healthy subjects) with a maximum plasma concentration (Cmax) of 8.9 nmol L−1 and resulting area under curve (AUC) of 139 nmol h L−1. Its absolute bioavailability is ∼30% and it exhibits concentration dependent plasma protein binding.10Extensive literature is available on LGN human PK/clinical studies, performed either alone7,11–16 or in combination17–21 with various other drugs. However, to the best of our knowledge, scientific reports on the optimized bioanalytical method for measuring LGN with full validation while addressing essential method details required for reproducing the results is lacking. These studies reporting respective LC-MS/MS methods provide only incomplete methodological details (refer to Section: 3.4) for linagliptin determination in human plasma samples16–18,20 or in both human plasma and urine samples12–15,19,21 with 13C3 labelled IS.12–16,18–21 One of these methods,15 simultaneously measured both LGN and the main inactive metabolite of LGN (CD 1790) in a single analytical run. For the measurement of LGN, these methods vary in sensitivity (range: 0.049 to 0.25 ng mL−1, 1 ng mL−1 of LGN = 2.116 nM (ref. 19 and 20)), upper limit of calibration curve (range: 49.14 to 250 ng mL−1), volume of plasma sample (range: 50 μL to 150 μL), retention time and run time (50 min and 70 min respectively with gradient elution), and all of these methods adopted the expensive Solid Phase Extraction (SPE) technique for sample purification. Further it is important to note that none of these methods neither comprehensively reported method validation experiments with results nor followed the latest regulatory guidelines for method validation.22–24
It is essential to establish a reliable bioanalytical method with complete validation for PK and bioequivalence [BE] in a study intended for regulatory submission. Therefore in view of the unavailability of sufficient literature for LGN estimation in biological samples, we aimed to develop a simple, sensitive, selective, high throughput and reliable bioanalytical method with full quality validation conducted as per the most recent version of regulatory guidelines.22–24
2 Experimental
2.1 Materials and reagents
Both LGN working standard (purity, 98.26%) and LGN-d4, IS (purity, 98.39%) (Fig. 1), were procured from Clearsynth Labs Ltd. (Mumbai, India). Methanol, water, ethyl acetate [HPLC grade], ammonium formate (extra pure grade), ammonia solution and formic acid [EMPARTA grade] was procured from Merck specialities Pvt. Ltd. (Mumbai, India). K2EDTA (dipotassium ethylene diamine tetra-acetic acid) healthy human control plasma (including the haemolyzed and lipemic nature) was obtained from Deccan Pathology Labs, Hyderabad.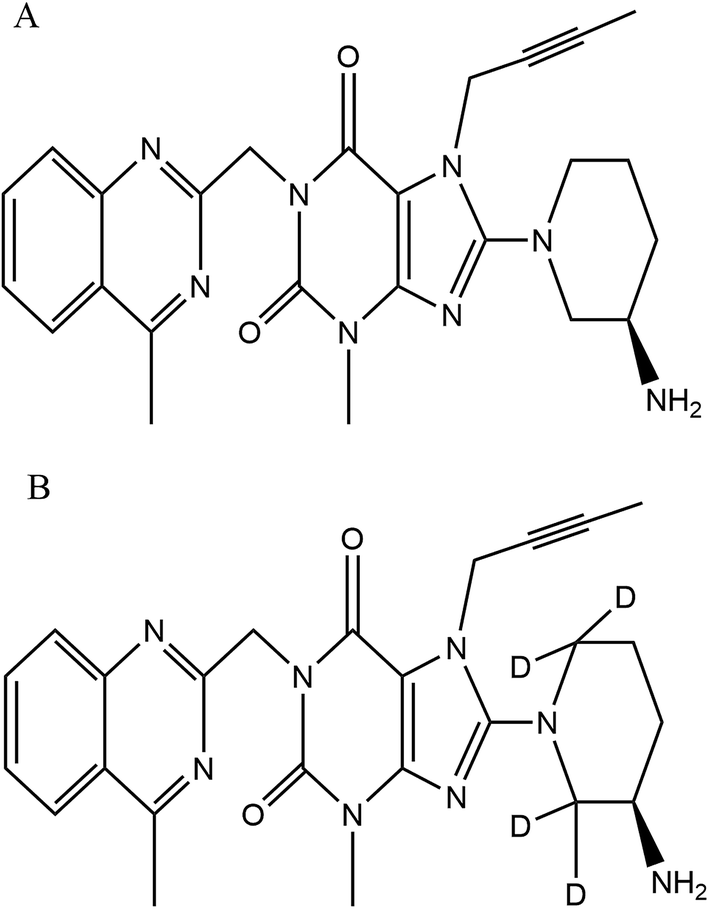 | ||
| Fig. 1 Chemical structure of (A) linagliptin, molecular weight: 472.54 g mol−1 and (B) linagliptin-d4, molecular weight: 476.57 g mol−1. | ||
2.2 Preparation of mobile phase buffer and stock solutions
0.31 g of ammonium formate (mobile phase buffer) was weighed into a reagent bottle containing 500 mL of HPLC water and mixed before filtration with a 0.22 μ filter.The primary stock solutions (PSS) of LGN and LGN-d4 were prepared in methanol (0.5 mg mL−1 and 0.4 mg mL−1 respectively) and stored in a refrigerator at 2–8 °C. When required, freshly prepared ISS (intermediate stock solutions) from PSS, using diluent [mixture of methanol–water (50:50, v/v)] at 50000 ng mL−1 for LGN and at 4000 ng mL−1 for IS. CC/QC spiking solutions were prepared in appropriate concentration ranges from the analyte ISS using same diluent.
2.3 Preparation of calibration curve standards (CC’s) and quality control samples (QC’s)
8 CC standards [50.3 (LLOQ-lower limit of quantitation), 114.5, 318.0, 1325.3, 4016.2, 6693.8, 10298.1 and 12115.5 pg mL−1 (ULOQ-upper limit of quantitation)] and 4 level QC samples [50.6 (LLOQ QC), 142.8 (low QC or LQC), 4760.3 (middle QC or MQC) and 9520.6 (high QC or HQC)] were prepared by spiking screened blank K2EDTA human plasma with respective CC/QC spiking solutions at 2% v/v. LGN-d4 working solution was prepared freshly (from PSS) as and when required at 40 ng mL−1 using the diluent mentioned above. Multiple CC’s, QC’s, standard blanks (blank plasma without spiking analyte and IS) and zero samples (blank plasma spiking with 50 μL LGN-d4 working solution during sample processing) in 0.4 mL quantity were aliquoted into pre-labelled polypropylene vials and stored in the deep freezer at (−70 ± 15 °C and −20 ± 5 °C) for conducting various validation parameters.
2.4 Instrumentation and LC-MS/MS conditions
The analysis was performed on UHPLC system [(Shimadzu, Kyoto, Japan) consisting of a binary pump – LC Nexera X2 LC 30 AD, degasser – DGU 20 A 5R, an auto sampler – Nexera X2 SIL 30ACMP and a column oven – Prominence CTO 20AC], interfaced with an API 4000 triple quadrupole (AB SCIEX, Singapore) tandem mass spectrometer, internally equipped with an electrospray ionization (Turbo v™) source. A nitrogen generator (Peak scientific, INFINITY 1031) was used to provide high purity nitrogen for the mass spectrometer. The chromatographic profile for the analyte and IS [RT at 1.72 (±0.30) and 1.71 (±0.30) min respectively with a run time of 3.5 min] was acquired with an isocratic mobile phase [a mixture of 10 mM ammonium formate:methanol [20:80 (v/v)] that was degassed ultrasonically for 5 min using an ultrasonic bath (HWASHIN TECH) and delivered at a flow rate of 0.5 mL min−1 on to a Gemini 3 μ, C18, 100 mm × 4.6 mm (Phenomenex) column, maintained at 35 ± 1 °C, while injecting a sample volume of 20 μL. Only 30% of the eluent from the column exit was split into the mass ionization source to facilitate MRM (multiple reaction monitoring) quantification (in positive ion mode) at the common optimized source and compound specific parameters, for both LGN and LGN-d4 with the resulting precursor to product ion transitions observed at m/z: 473.3/420.1 and 477.5/424.3 respectively (Fig. 2). The ion source temperature and spray voltage was maintained at 500 °C and 5500 V respectively and quadrupoles Q1 and Q3 were set on unit resolution with a dwell time of 200 ms for each mass transition. GS1 (nebulizer gas), GS2 (turbo gas), curtain and CAD (collision associated dissociation) gas source parameters were set at 40, 45, 40 and 6 psi, respectively. Compound dependent parameters, viz. DP (declustering potential), CE (collision energy), EP (entrance potential) CXP (collision cell exit potential), were set at 90, 31, 10 and 13 V respectively. Instrument management and data acquisition was performed using Analyst software version 1.6.2 (AB Sciex, Singapore).
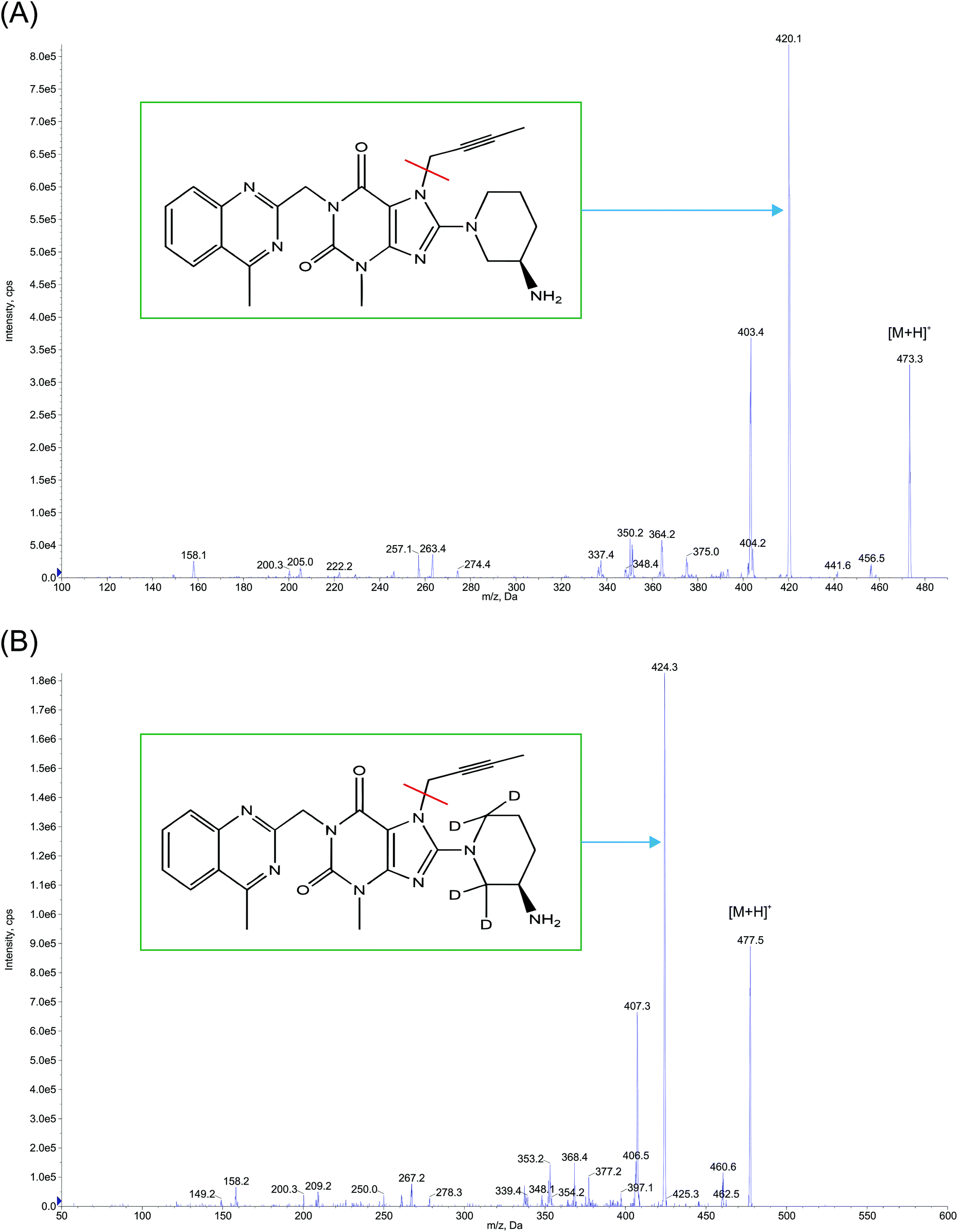 | ||
| Fig. 2 Product ion mass spectra of protonated (A) linagliptin (m/z: 473.3 → 420.1, scan range: 100–500 Da) and (B) linagliptin-d4 (m/z: 477.5 → 424.3, scan range: 50–600 Da) in ESI mode. | ||
2.5 Protocol for sample extraction procedure
A simple LLE (liquid–liquid extraction) method was followed for isolating both the analyte and the IS from the plasma sample. Each 0.3 mL aliquot from the CC standard, QC sample, standard blank, zero sample and unknown subject sample was spiked with 50 μL of LGN-d4 IS working solution (except for the standard blank sample) followed by vortex mixing and the addition of 0.4 mL extraction buffer [10% v/v ammonia solution in water]. The resulting sample content was first vortex mixed followed by the addition of 2.5 mL of the extraction solvent (ethyl acetate), then further mixed on a vibramax reciprocating shaker for 20 min at 2000 rpm before undergoing centrifugation for 10 min (3500 rpm at 5 °C) on an Eppendorf 5810 R (Eppendorf, Germany) centrifuge machine. Two (2.0) mL of the supernatant organic layer obtained post centrifugation was evaporated to dryness under nitrogen gas (Rapid 50, Crescent Scientific India) at 50 °C. The dried residue was reconstituted with 400 μL of the mobile phase (reconstitution solution) and the reconstituted sample, after vortex mixing was loaded into the autosampler vials of the UHPLC system for injection of 20 μL of solution onto the column.2.6 Method validation
Method validation was performed in accordance with the most recent version of the US-FDA, EMA and ANVISA guidelines.22–24 The precision (P) of the method was expressed in terms of % CV (coefficient of variation), calculated as the ratio of standard deviation/mean. Accuracy (A) was defined as: percent difference of mean observed value from nominal value, expressed as % nominal or calculated concentration/nominal concentration × 100. Eight point calibration standards and four level QC samples at appropriate level (refer, Section 2.3) in replicates were used for establishing the majority of the validation experiments described below.834.7 pg mL−1) equivalent to 1.6 times of ULOQ were diluted 5 fold with previously screened plasma and analyzed along with three P & A batches on a with-in and between-run basis.Within-run and between-run P & A was established where precision should lie within 20.0% CV for LLOQ QC and within 15% CV incase of other QC’s. The accuracy for LLOQ QC should be with in ±20.0% (from their nominal concentrations) and for other QC’s, it should be within ±15.0%. At least 67% of overall QC samples and 50% at each QC level should meet this criterion.
Stock solutions stability. Comprising of short term stock solution stability (STSSS) and long term stock solution stability (LTSSS) for the analyte (LLOQ and ULOQ level) and IS (working concentration level), by comparing the mean peak area of the stability stock with comparison stock, determined by injecting six replicates.
In the case of STSSS, the primary stock kept at 25 ± 5 °C was assigned as stability stock and the corresponding stock retrieved from refrigerated conditions (2–8 °C) was assigned as the comparison stock. For LTSSS, the respective stocks prepared from fresh weighing was designated as comparison stock vs. the corresponding stock which was stored at 2–8 °C in the refrigerator and retrieved for analysis, was designated as stability stock.
The stock solution was considered stable if the mean peak area of the respective stability sample was within ±10% (90–110%) from the mean peak area of the corresponding comparison sample, while meeting the precision criteria of ≤15% for both stability and comparison samples.
Stability studies in the plasma sample. Bench top stability (BTS), freeze thaw stability (FTS), dry extract stability (DES), stability of extract (SE), long term stability (LTS), stability of analyte in K2EDTA blood (SAB) were conducted at LQC and HQC level in 6 replicates, where the respective stability sample’s mean back calculated concentrations (calculated using freshly spiked CC’s) were compared to that of nominal concentrations (excepting SAB) with common P & A acceptance criteria of ±15.0%.
BTS samples were kept on the bench at ambient temperature (25 ± 5 °C) before processing for analysis. FTS was determined after freezing the samples in deep freezer at −70 ± 15 °C and −20 ± 5 °C for a minimum period of 12 hours and then completely thawed at ambient temperature in a water bath. DES was conducted (since sample processing involve evaporation step) after storing dried extract samples in refrigerator (2–8 °C). SE was performed (after immediate reconstitution with mobile phase) at ambient (25 ± 5 °C) and at refrigerator (2–8 °C) temperature. LTS was evaluated for samples stored in the deep freezer at −20 ± 5 °C and −70 ± 15 °C.
SAB was studied separately at ambient (25 ± 5 °C) and ice water bath (10 ± 3 °C) conditions, comparing the mean peak area ratio (analyte/IS) of these stability samples with freshly prepared comparison samples.
2.7 Application of the method to biological samples
Successfully validated method was subjected to measure plasma linagliptin concentrations of 12 healthy adult male Indian subjects whose samples previously collected for PK study (conducted in compliance with the relevant laws and as per ICH-GCP norms after approval from the Maarg independent ethics committee, Secunderabad, India. Informed consent was obtained from the study participants) after administration of single 5 mg tablet under fasting conditions.3 Results and discussion
3.1 Method development
To fulfill the objective of developing a simple, sensitive, selective, rapid, and reliable bioanalytical method of LGN estimation in human plasma using the LC-MS/MS method, a thorough review of the collective literature on LGN; from physicochemical,8,10,25 pharmacokinetic aspects8,10 and reported bioanalytical methods7,11–21 contributed a vital role towards the development process which included a selection of appropriate chromatographic-mass spectrometric conditions and extraction protocols before optimization.:water 80:20 v/v and injected at a flow rate of 10.0 μL min−1 using an infusion pump) of LGN and LGN-d4 into the ionization source, to determine their respective Q1 mass of the parent ion and Q3 mass of the product ion (Fig. 2) and to generate the optimal source and compound specific parameters as explained in Section 2.4. For the analyte and IS, mass parameters were tested initially in both positive and negative ionization modes. MRM method in positive ion mode was implemented in the optimized method since it gave a consistent mass response where the observed precursor to product ion mass transition of LGN matches with the earlier literature.15:2 mM amm. (ammonium) acetate pH – 4.0 (85:15 and 80:20% v/v, with corresponding flow rates (FR) of 0.8 and 0.4 mL min−1), MEOH:0.1% formic acid (80:20%v/v), FR:0.5 mL min−1, MEOH:10 mM amm. formate pH-4.00 (80:20% v/v), FR: 0.6 mL min−1, acetonitrile:5 mM amm. acetate pH-4.01 (85:15% v/v), FR: 0.8 mL min−1. The best chromatographic and mass optimization (Fig. 3) achieved (while maintaining the consistent column back pressure between 1400 and 1600 psi until 12 hours continuous analysis) with Gemini 4.6 × 100 mm, 3 μ (Phenomenex) and MEOH:10 mM ammonium formate (80:20% v/v), delivered at a flow rate of 0.5 mL min−1 with a sample injection volume of 20 μL. Only C18 columns were tested based on prior knowledge13,15 and also the logP of the linagliptin molecule.25
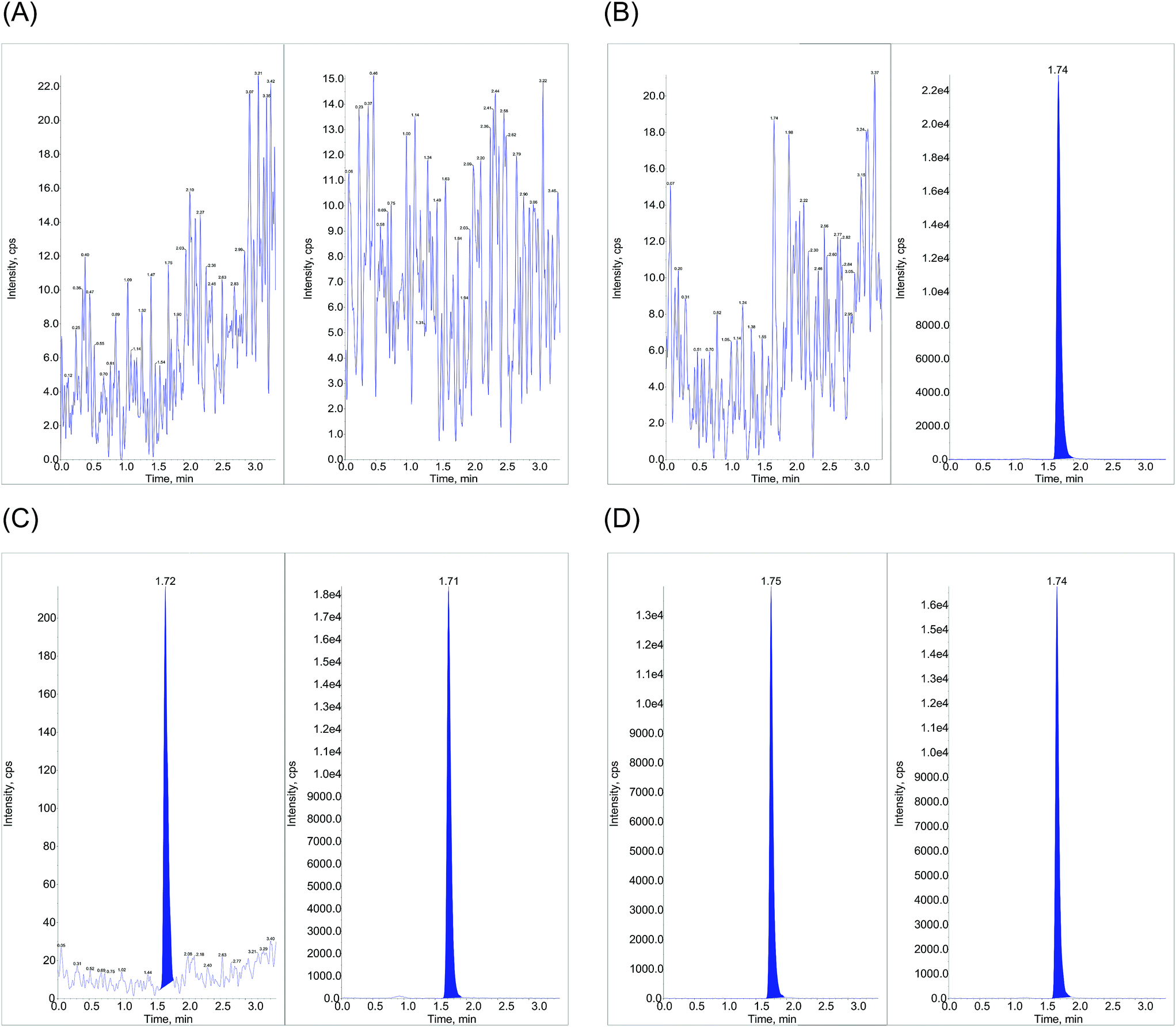 | ||
| Fig. 3 UHPLC-MS/MS/MRM ion extracted plasma chromatograms for (A) standard blank (without spiking analyte and IS) (B) zero sample (spiked with only IS – 50 μL of 40 ng mL−1 solution) (C) LLOQ (50.3 pg mL−1) and (D) post dose subject sample (spiked with IS – 50 μL of 40 ng mL−1 solution) at 2.5 hours. In each figure, the left panel represents linagliptin and the right panel shows linagliptin-d4. | ||
Though the SPE technique was adopted in some of earlier reports12,13,15–20 it was not given priority as this method, in comparison with LLE, is time consuming and requires expensive materials.31 Therefore, in the present work a simple LLE method, as explained under Section 2.5, was implemented. LLE has the merits of minimizing the experimental cost, introducing a clean sample for spectral analysis in general32 and a short sample processing time along with very good and consistent overall recoveries of the analyte and IS (not less than 70.9%) while confirming the absence of any significant matrix effects. Various extraction solvents investigated for recovery experiments include, ethyl acetate:N-hexane in different volume ratio’s, 100% TBME in presence of different buffer conditions, acid-0.1 N HCl, base-25 mM K2HPO4. 100% ethyl acetate when used in the presence of a base – 5% ammonia solution in water gave comparatively low recovery, where as 20% ammonia solution gave inconsistent recovery at LQC level.
3.2 Assay performance and validation
| QC sample (n = 6) | Post extracted sample peak area | Extracted sample peak area | Mean recoverya, % | Global recoveryb, % | ||
|---|---|---|---|---|---|---|
| (Mean ± SD) | % CV | (Mean ± SD) | % CV | |||
a 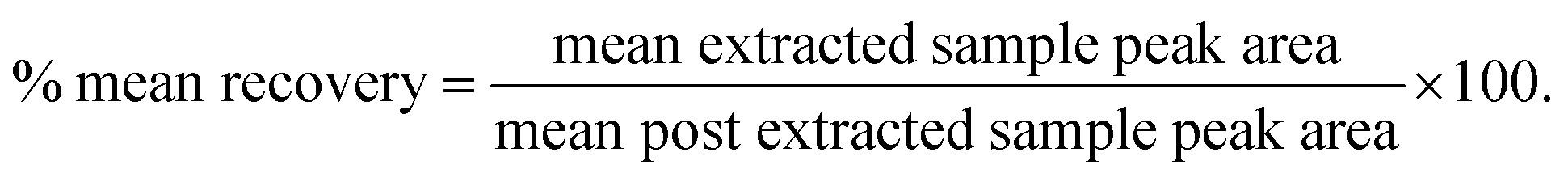 b Average mean recovery of 3 QC’s. Abbreviations: LQC: low quality control, MQC: middle quality control, HQC: high quality control, SD: standard deviation, % CV: percentage coefficient of variation, n = 6: total number of observations. b Average mean recovery of 3 QC’s. Abbreviations: LQC: low quality control, MQC: middle quality control, HQC: high quality control, SD: standard deviation, % CV: percentage coefficient of variation, n = 6: total number of observations. |
||||||
| LQC | 2496.7 ± 70.3 | 2.8 | 1635.0 ± 120.9 | 7.4 | 65.5 | |
| MQC | 85163.5 ± 2310.9 |
2.7 | 59773.8 ± 3373.0 |
5.6 | 70.2 | 71.0 |
| HQC | 161749 ± 6952.2 |
4.3 | 124999.8 ± 9805.7 |
7.8 | 77.3 | |
| Plasma lot no. | MF | IS normalized MF at HQC level (9520.6 pg mL−1) | MF | IS normalized MF at LQC level (142.8 pg mL−1) | ||
|---|---|---|---|---|---|---|
| HQC | IS | LQC | IS | |||
Abbreviations: LQC: low quality control, HQC: high quality control, % CV: percentage coefficient of variation, SD: standard deviation, MF: matrix factor, IS: internal standard, L: lipemic plasma, H: hemolytic plasma, 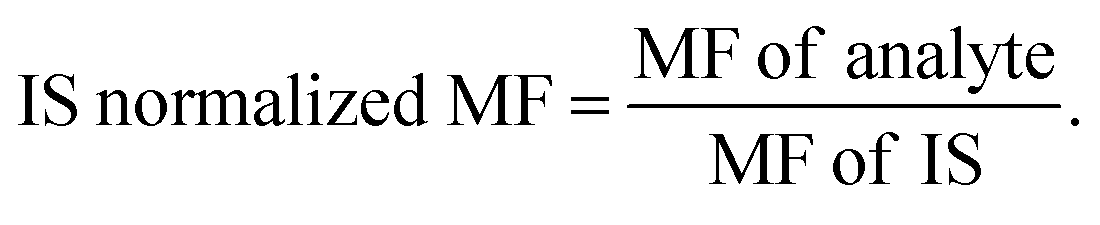 |
||||||
| 1 | 1.19 | 1.23 | 0.97 | 1.14 | 1.13 | 1.01 |
| 2 | 1.16 | 1.19 | 0.97 | 1.17 | 1.11 | 1.05 |
| 3 | 1.06 | 1.07 | 0.99 | 0.95 | 0.98 | 0.97 |
| 4 | 1.07 | 1.06 | 1.01 | 1.11 | 1.03 | 1.08 |
| 5 | 1.04 | 1.03 | 1.01 | 1.00 | 0.96 | 1.04 |
| 6 | 1.04 | 1.00 | 1.04 | 0.99 | 1.00 | 1.00 |
| 7 (L) | 1.01 | 1.03 | 0.98 | 1.05 | 1.00 | 1.06 |
| 8 (L) | 1.04 | 1.05 | 0.99 | 0.98 | 1.03 | 0.95 |
| 9 (H) | 0.98 | 0.99 | 0.99 | 1.01 | 1.02 | 1.00 |
| 10 (H) | 1.05 | 1.05 | 1.00 | 0.98 | 0.98 | 1.00 |
| Mean | 1.06 | 1.07 | 0.99 | 1.04 | 1.02 | 1.01 |
| SD | 0.022 | 0.042 | ||||
| % CV | 2.2 | 4.1 | ||||
115.5 pg mL−1, using a linear regression model with 1/x2 weighing produced best result (r = 0.9994). For CC standards observed P & A were 0.9 to 3.2% CV and −5.3 to 2.8% nominal respectively. Refer to Table 3 for a summary of the results on P & A of the method.
| Nominal conc. of QC’s (pg mL−1) | Within-run (n = 6) | Between-run (n = 18) | ||||
|---|---|---|---|---|---|---|
| Conc. found meana ± SD (pg mL−1) | Precision (% CV) | Accuracy (%) | Conc. found meanb ± SD (pg mL−1) | Precision (% CV) | Accuracy (%) | |
| a Mean of six replicate sample observations from one analytical validation run.b Mean of eighteen replicate sample observations from three separate analytical validation runs. n = total number of observations. Abbreviations: LLOQ QC: lower limit of quantitation quality control, LQC: low quality control, MQC: middle quality control, HQC: high quality control, % CV: percentage coefficient of variation, SD: standard deviation. | ||||||
| LLOQ QC (50.6) | 47.7 ± 4.1 | 8.6 | 94.3 | 47.1 ± 3.2 | 7.0 | 93.1 |
| LQC (142.8) | 123.8 ± 7.2 | 5.8 | 86.7 | 130.1 ± 7.8 | 6.0 | 91.2 |
| MQC (4760.3) | 4276.05 ± 48.5 | 1.1 | 89.8 | 4440.8 ± 149.9 | 3.4 | 93.3 |
| HQC (9520.6) | 8695.9 ± 95.3 | 1.1 | 91.3 | 9100.3 ± 325.8 | 3.6 | 95.6 |
Stability of analyte and IS in stock solutions. For analyte and IS, short term stock solution stability was established for 07 hours 40 min and 06 hours 16 min respectively. Their long term stock solution stability was recorded for the duration of 19 days, 21 hours and 2 days, 23 hours respectively. Mean peak area of stability samples was in the range of 91.3 to 103.8% in relation to the comparison samples. Observed precision range among stability and comparison samples was 1.0 to 9.1%.
Stability of analyte in plasma sample. Designed studies met predefined acceptance criteria. Refer to Table 4 for a summary of results.
| Condition of storage & stability duration | QC sample | Nominal concentration (pg mL−1) | Calculated concentration (mean ± SD) | Precision (% CV) | Accuracy (%) | % changea |
|---|---|---|---|---|---|---|
a  Abbreviations: BTS: bench top stability, FTS: freeze–thaw stability, DES: dry extract stability, SE: stability of extract, LTS: long term stability, LQC: low quality control, HQC: high quality control, SD: standard deviation, % CV: percentage coefficient of variation. Abbreviations: BTS: bench top stability, FTS: freeze–thaw stability, DES: dry extract stability, SE: stability of extract, LTS: long term stability, LQC: low quality control, HQC: high quality control, SD: standard deviation, % CV: percentage coefficient of variation. |
||||||
| BTS (25 ± 5 °C, 7.15 h) | LQC | 142.8 | 141.0 ± 6.6 | 4.7 | 98.7 | −1.26 |
| HQC | 9520.6 | 9631.3 ± 458.9 | 4.8 | 101.2 | 1.16 | |
| FTS (after 5 cycle, −70 ± 15 °C) | LQC | 142.8 | 148.3 ± 7.2 | 4.9 | 103.8 | 3.85 |
| HQC | 9520.6 | 9539.8 ± 249.3 | 2.6 | 100.2 | 0.20 | |
| FTS (after 5 cycle, −20 ± 5 °C) | LQC | 142.8 | 144.9 ± 6.0 | 4.1 | 101.5 | 1.47 |
| HQC | 9520.6 | 9421.4 ± 128.4 | 1.4 | 99.0 | −1.04 | |
| DES (2–8 °C, 26.39 h) | LQC | 142.8 | 136.0 ± 6.1 | 4.5 | 95.2 | −4.76 |
| HQC | 9520.6 | 9686.9 ± 65.3 | 0.7 | 101.7 | 1.74 | |
| SE (25 ± 5 °C, 4.14 h) | LQC | 142.8 | 125.8 ± 3.1 | 2.5 | 88.1 | −11.90 |
| HQC | 9520.6 | 9073.8 ± 199.6 | 2.2 | 95.3 | −4.69 | |
| SE (2–8 °C, 26.39 h) | LQC | 142.8 | 132.6 ± 5.9 | 4.4 | 92.9 | −7.14 |
| HQC | 9520.6 | 9122.5 ± 223.1 | 2.4 | 95.8 | −4.18 | |
| LTS (−70 ± 15 °C, 118 days) | LQC | 142.8 | 140.2 ± 6.1 | 4.4 | 98.2 | −1.82 |
| HQC | 9520.6 | 9247.2 ± 157.9 | 1.7 | 97.1 | −2.87 | |
| LTS (−20 ± 5 °C, 118 days) | LQC | 142.8 | 153.0 ± 8.1 | 5.3 | 107.7 | 7.14 |
| HQC | 9520.6 | 10276.6 ± 213.9 |
2.1 | 108.5 | 7.94 | |
Stability of analyte in K2EDTA blood. LQC and HQC level of stability samples kept at ambient and refrigerator conditions for a period of 02 hours 08 minutes did not deviate by more than 4.1% from freshly prepared samples.
3.3 Application
The fully validated method was applied to PK study samples with the successful incurred sample reanalysis in a separate batch where the % variability was within 20% for more than 90% samples. A representative linear plasma concentration vs. time profile for one of the subjects was shown in Fig. 4.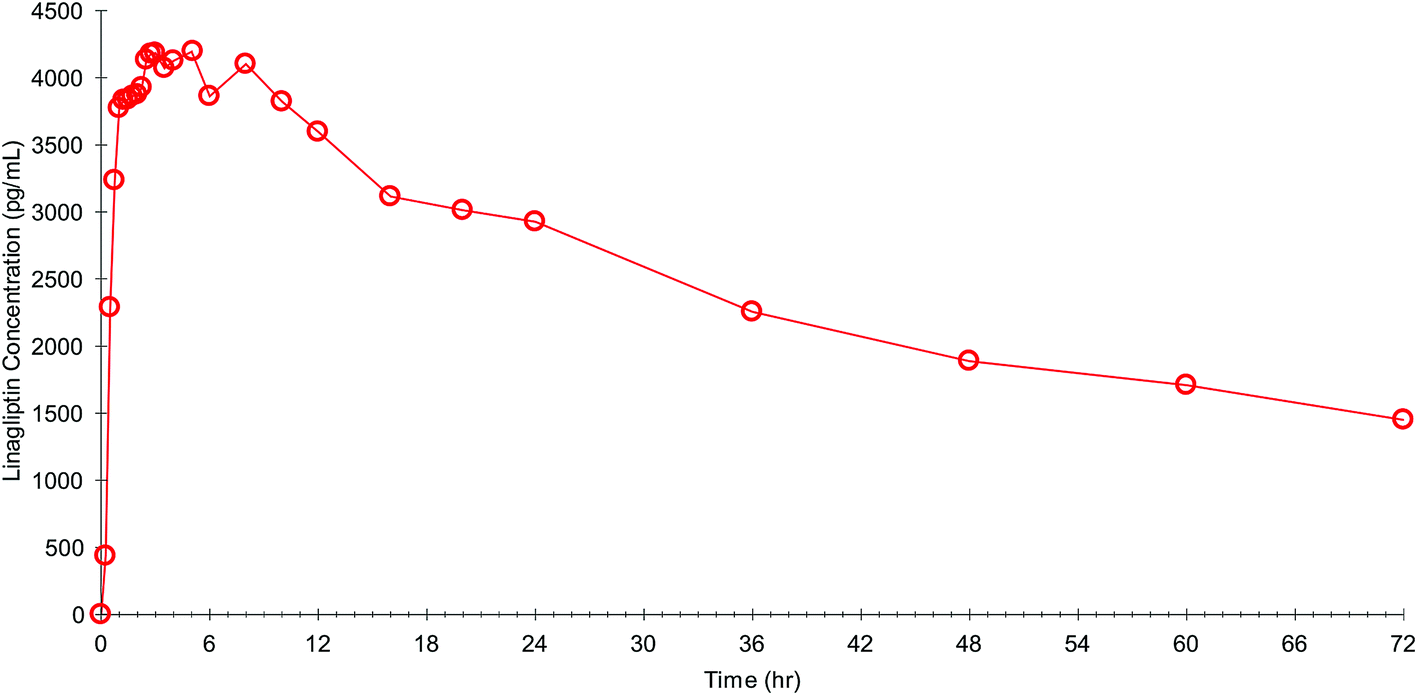 | ||
| Fig. 4 A representative plasma concentration vs. time profile of one subject following administration of single 5 mg tablet of linagliptin. | ||
3.4 Comparison of proposed method with previous published reports
Table 5 highlight the merits of our proposed method (PM) in comparison with the existing literature. Common unfavorable attributes of these published methods12–21 which were not designed in line with the latest regulatory requirements, did not report the detailed procedures for (1) optimization of LC and MS/MS conditions (2) sample cleanup by SPE (3) various validation parameters to prove the reliability of the method along with their results. Furthermore, these methods employed (1) 13C3 LGN as internal standard which is very difficult to obtain from authorized commercial sources and custom synthesis can be very expensive and time consuming (2) an expensive and time consuming SPE protocol for sample clean up. (3) Very wide calibration range, most importantly with respect to the upper limit (50 to 250 ng mL−1). This is abnormally very high to reflect the low concentrations of LGN from the clinical studies, especially when conducted on approved oral doses of the 2.5 mg (multi drug combination) and 5 mg tablet respectively.8,10 As per the regulatory requirements, and from a scientific perspective, in order to appropriately characterize the PK profile, it is mandatory to choose an appropriate calibration range based on the expected concentrations of the analyte of interest in clinical study programs.22–24 Several drawbacks were found even in the latest LC-MS/MS method reported by Shafi S. S. M. et al.33 which include: in appropriated CC and QC range (10 ng mL−1 to 5000 ng mL−1 and 10 ng mL−1 to 4000 ng mL−1 receptively) used in method development and validation, the insensitive characteristic of the method with validated LLOQ (10 ng mL−1) which was almost 2.4 times higher than the reported Cmax [8.9 nmol L−1 or 4.2 ng mL−1 (1 ng mL−1 of LGN = 2.116 nM)] of LGN at a maximum oral dose of 5 mg, usage of large plasma sample volume (450 μL), incomplete method validation experiments in line with latest regulatory guidelines, usage of normal IS instead of stable labeled IS etc. However there were some similar differences between their method and our proposed method with respect to the extraction protocol, usage of C18 column and sample run time. Our proposed method, except comparatively use high plasma volume than the previous reports, had superior results in all other aspects while fulfilling our aim of establishing a simple, rapid, reliable, sensitive and selective method for estimating LGN in human plasma. Furthermore, the LOQ of our method at pg mL−1 (50.3 pg mL−1) could help to address expected lower plasma levels even from low dose linagliptin (dose lower than 5 mg) studies.| S.N | Method | IS | Biological matrixa | EM | SV (μL) | Cal. rangeb (ng mL−1) | RTPS (min) | RT (min) | Reporting full method details (yes/no) | Reporting of MV exp. with results (yes/no) | Followed latest reg. guid.c | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Most of these published methods analysed LGN in both human plasma and urine sample using different level of calibration range and calibration range in this table pertains only to LGN in human plasma.b Calibration range given in published reports in nM L−1 converted to ng mL−1 (1 ng mL−1 of LGN = 2.116 nM (ref. 19 and 20)).c US FDA (2013), EMEA (2011) and ANVISA (2012).d Inactive main metabolite (CD 1750) of LGN also estimated.e SPE procedure was not reported in detail, – not reported, PR: partial reporting. Abbreviations: S.N: serial number, IS: internal standard, EM: extraction method, SV: sample volume, Cal. range: validated calibration curve range, RTPS: run time per sample, RT: retention time of LGN (linagliptin), MV: method validation, exp: experiment, reg. guid.: regulatory guideline, Ref.: reference, SPE: solid phase extraction, LLE: liquid–liquid extraction. | ||||||||||||
| 1 | LC-MS/MS | 13C3 LGN | Human K2EDTA plasma | SPEe | 50 | 0.25 to 250 | — | — | No | No | No | 12 and 13 |
| 2 | LC-MS/MS | 13C3 LGN | Human plasma | — | — | — | — | — | No | No | No | 14 |
| 3 | LC-MS/MS | 13C3 LGN & 13C3 CD 1750d | Human plasma | SPEe | 150 | 0.049 to 49.14 | 70 | 50 | No | No | No | 15 |
| 4 | LC-MS/MS | 13C3 LGN | Human EDTA plasma | SPEe | 150 | 0.05 to 50 | — | — | No | No | No | 16–20 |
| 5 | LC-MS/MS | — | Human EDTA plasma | — | — | — | — | — | — | — | — | 21 |
| 6 | LC-MS/MS | Telmisartan | Human plasma | LLE | 450 | 10.0 to 5000 | 3.0 | 1.42 | No | PR | No | 33 |
| 7 | UHPLC-MS/MS | LGN-d4 | Human K2EDTA plasma | LLE | 300 | 0.05 to 12.1 | 3.5 | 1.75 | Yes | Yes | Yes | PM |
4 Conclusions
To the best of our knowledge, this is the first time a sensitive, rapid, rugged, selective and simple bioanalytical method was developed using UHPLC-MS/MS in human plasma with successful full validation as per the combined requirements of the most recent regulatory guidelines. The validated method was further tested to analyse PK study samples with successful incurred sample reanalysis.All potential features described in Section 3.4 collectively make this method attractive for high throughput analytical demands from large cohort clinical, PK and BE studies of linagliptin, especially for regulatory submission and it is further expected to advance new clinical study programs on linagliptin.
Conflicts of interest
Authors disclose no conflicts.Acknowledgements
Authors greatly acknowledge management of Jeevan Scientific Technologies Ltd, for providing the infrastructure facility and resources to carry out this work. Authors would like to express their gratitude to the Chief Librarian of IICT (Indian Institute of Chemical Technology), Hyderabad, India for facilitating library access to collect literature.References
- IDF Diabetes, 7th edn, 2015, http://www.diabetesatlas.org, accessed April 2016.
- C. F. Deacon, Dipeptidyl peptidase-4 inhibitors in the treatment of type 2 diabetes: a comparative review, Diabetes, Obes. Metab., 2011, 13, 7–18 CrossRef CAS PubMed.
- M. Lehrke, N. Marx, S. Patel, T. Seck, S. Crowe, K. Cheng, M. von Eynatten and O. E. Johansen, Safety and tolerability of linagliptin in patients with type 2 diabetes: a comprehensive pooled analysis of 22 placebo-controlled studies, Clin. Ther., 2014, 36, 1130–1146 CrossRef CAS PubMed.
- H. Fuchs, F. Runge and H.-D. Held, Excretion of the dipeptidyl peptidase-4 inhibitor linagliptin in rats is primarily by biliary excretion and P-gp-mediated efflux, Eur. J. Pharm. Sci., 2012, 45, 533–538 CrossRef CAS PubMed.
- S. Del Prato, M.-R. Taskinen, D. R. Owens, M. von Eynatten, A. Emser, Y. Gong, S. Chiavetta, S. Patel and H.-J. Woerle, Efficacy and safety of linagliptin in subjects with type 2 diabetes mellitus and poor glycemic control: pooled analysis of data from three placebo-controlled phase III trials, Journal of Diabetes and its Complications, 2013, 27, 274–279 CrossRef PubMed.
- T. Karagiannis, P. Paschos, K. Paletas, D. R. Matthews and A. Tsapas, Dipeptidyl peptidase-4 inhibitors for treatment of type 2 diabetes mellitus in the clinical setting: systematic review and meta-analysis, Br. Med. J., 2012, 344, e1369 CrossRef PubMed.
- U. Graefe-Mody, P. Rose, S. Retlich, A. Ring, L. Waldhauser, R. Cinca and H. J. Woerle, Pharmacokinetics of linagliptin in subjects with hepatic impairment, Br. J. Clin. Pharmacol., 2012, 74, 75–85 CrossRef CAS PubMed.
- U. Graefe-Mody, S. Retlich and C. Friedrich, Clinical pharmacokinetics and pharmacodynamics of linagliptin, Clin. Pharmacokinet., 2012, 51, 411–427 CrossRef CAS PubMed.
- E. D. Deeks, Linagliptin: a review of its use in the management of type 2 diabetes mellitus, Drugs, 2012, 72, 1793–1824 CrossRef CAS PubMed.
- Tradjenta® (linagliptin) tablets full prescribing information, http://docs.boehringer-Ingelheim.com/Prescribing%20Information/PIs/Tradjenta/Tradjenta.pdf, accessed April, 2016.
- C. Friedrich, S. Glund, D. Lionetti, C. J. Kissling, J. Righetti, S. Patel, U. Graefe-Mody, S. Retlich and H. J. Woerle, Pharmacokinetic and pharmacodynamic evaluation of linagliptin in African American patients with type 2 diabetes mellitus, Br. J. Clin. Pharmacol., 2013, 76, 445–454 CrossRef CAS PubMed.
- A. Sarashina, S. Sesoko, M. Nakashima, N. Hayashi, A. Taniguchi, Y. Horie, E. U. Graefe-Mody, H.-J. Woerle and K. A. Dugi, Linagliptin, a dipeptidyl peptidase-4 inhibitor in development for the treatment of type 2 diabetes mellitus: a phase I, randomized, double-blind, placebo-controlled trial of single and multiple escalating doses in healthy adult male Japanese subjects, Clin. Ther., 2010, 32, 1188–1204 CrossRef CAS PubMed.
- S. Hüttner, E. U. Graefe-Mody, B. Withopf, A. Ring and K. A. Dugi, Safety, tolerability, pharmacokinetics, and pharmacodynamics of single oral doses of BI 1356, an inhibitor of dipeptidyl peptidase 4, in healthy male volunteers, J. Clin. Pharmacol., 2008, 48, 1171–1178 CrossRef PubMed.
- C. Friedrich, X. Shi, P. Zeng, A. Ring, H. Juergen Woerle and S. Patel, Pharmacokinetics of single and multiple oral doses of 5 mg linagliptin in healthy Chinese volunteers, Int. J. Clin. Pharmacol. Ther., 2012, 50, 889–895 CrossRef CAS PubMed.
- S. Blech, E. Ludwig-Schwellinger, E. Ulrike Graefe-Mody, B. Withopf and K. Wagner, The metabolism and disposition of the oral dipeptidyl peptidase-4 inhibitor, linagliptin, in humans, Drug Metab. Dispos., 2010, 38, 667–678 CrossRef CAS PubMed.
- U. Graefe-Mody, T. Giessmann, A. Ring, M. Iovino and H.-J. Woerle, A randomized, open-label, crossover study evaluating the effect of food on the relative bioavailability of linagliptin in healthy subjects, Clin. Ther., 2011, 33, 1096–1103 CrossRef CAS PubMed.
- C. Friedrich, A. Ring, T. Brand, R. Sennewald, E. U. Graefe-Mody and H. J. Woerle, Evaluation of the pharmacokinetic interaction after multiple oral doses of linagliptin and digoxin in healthy volunteers, Eur. J. Drug Metab. Pharmacokinet., 2011, 36, 17–24 CrossRef CAS PubMed.
- S. Buschke, A. Ring, C. Friedrich, K. Matzmann and T. Meinicke, Linagliptin fixed-dose combination with metformin is bioequivalent to co-administration of linagliptin and metformin as individual tablets, Int. J. Clin. Pharmacol. Ther., 2014, 52, 537–548 CAS.
- E. U. Graefe-Mody, S. Padula, A. Ring, B. Withopf and K. A. Dugi, Evaluation of the potential for steady-state pharmacokinetic and pharmacodynamics interactions between the DPP-4 inhibitor linagliptin and metformin in healthy subjects, Curr. Med. Res. Opin., 2009, 25, 1963–1972 CrossRef CAS PubMed.
- U. Graefe-Mody, P. Rose, A. Ring, K. Zander, M. Lovino and H.-J. Woerle, Assessment of pharmacokinetic interaction between the novel DPP-4 inhibitor linagliptin and a sulfonylurea, glyburide in healthy subjects, Drug Metab. Pharmacokinet., 2011, 26, 123–129 CrossRef CAS PubMed.
- C. Friedrich, K. Metzmann, P. Rose, M. Mattheus, S. Pinnetti and H. J. Woerle, A randomized, open-label, crossover study to evaluate the pharmacokinetics of empagliflozin and linagliptin after coadministration in healthy male volunteers, Clin. Ther., 2013, 35, A33–A42 CrossRef CAS PubMed.
- FDA guidancefor industry bioanalytical method validation (draft guidance), US department of health and human services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), September 2013.
- European Medicines Agency guideline on bioanalytical method validation adopted by Committee for Medicinal Products for Human Use (CHMP), 21st July 2011.
- ANVISA resolution no. 27 of 17/05/12, Federal Official Gazette (DOU).
- DrugBank database, http://www.drugbank.ca/drugs/DB08882, accessed March, 2016.
- J. F. Bower, J. B. McClung, C. Watson, T. Osumi and K. Pastre, Recommendations and best practices for reference standards and reagents used in bioanalytical method validation, AAPS J., 2014, 16, 352–356 CrossRef CAS PubMed.
- H. E. Zaazaa, E. S. Elzanfaly, A. T. Soudi and M. Y. Salem, Development and validation of an ultra-performance liquid chromatography method coupled with tandem mass spectrometry for determination of alizapride in human plasma, RSC Adv., 2015, 5, 76377–76382 RSC.
- T. A. Wani, Highly sensitive ultra-performance liquid chromatography-tandem mass spectrometry method for the determination of abiraterone in human plasma, Anal. Methods, 2013, 5, 3693–3699 RSC.
- S. Chen and A. Kord, Theoretical and experimental comparison of mobile phase consumption between ultra-high-performance liquid chromatography and high performance liquid chromatography, J. Chromatogr. A, 2009, 1216, 6204–6209 CrossRef CAS PubMed.
- C. Bylda, R. Thiele, U. Kobold and D. A. Volmer, Recent advances in sample preparation techniques to overcome difficulties encountered during quantitative analysis of small molecules from biofluids using LC-MS/MS, Analyst, 2014, 139, 2265–2276 RSC.
- J. R. Patel, T. M. Pethani, A. N. Vachhani, N. R. Sheth and A. V. Dudhrejiya, Development and validation of bioanalytical method for simultaneous estimation of ramipril and hydrochlorothiazide in human plasma using liquid chromatography-tandem mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 970, 53–59 CrossRef CAS PubMed.
- R. Muppavarapu, S. Guttikar, M. Rajappan, K. Kamarajan and R. Mullangi, Sensitive LC-MS/MS-ESI method for simultaneous determination of montelukast and fexofenadine in human plasma: application to a bioequivalence study, Biomed. Chromatogr., 2014, 28, 1048–1056 CrossRef CAS PubMed.
- S. S. Mahamad Shafi, A. Begum and N. D. V. R. Saradhi, Bioanalytical method development and validation of linagliptin in plasma through LC-MS/MS, Int. J. Bioassays, 2014, 3, 3146–3151 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |