
Constructing novel Ag nanoparticles anchored on MnO2 nanowires as an efficient visible light driven photocatalyst†
K. Saravanakumara,
V. Muthuraj*a and
S. Vadivelb
aDepartment of Chemistry, VHNSN College, Virudhunagar-626 001, Tamil Nadu, India. E-mail: muthuraj75@gmail.com; Tel: +91 9940965228
bDepartment of Chemistry, NGM College, Pollachi, Tamilnadu, India
First published on 17th June 2016
Abstract
In this study, novel Ag@MnO2 nanowires were constructed, using a one step hydrothermal method, which exhibited excellent efficiency towards the photodegradation of organic contaminants under visible-light driven irradiation. The resulting Ag@MnO2 nanowires were systematically characterized using various spectroscopic and microscopic techniques. Morphological characterizations show that the Ag nanoparticles are well anchored on the surface of the MnO2 nanowires. The N2 adsorption/desorption studies revealed that the as prepared photocatalyst possesses mesoporosity. The optical properties and energy band gap structures were studied using UV-visible diffuse reflectance spectroscopy. The photocatalytic activity enhancement of the Ag@MnO2 (5%) nanowires could be ascribed to the efficient separation of the photogenerated electron–hole pairs compared to other Ag@MnO2 nanowires and pure MnO2. The possible photocatalytic mechanism was proposed for this enhanced charge separation performance using photoluminescence spectra analysis, electrochemical impedance spectroscopy (EIS) spectra and photocurrent density. Furthermore, photocatalytic mechanism investigations demonstrate that ˙OH and O2˙− play a key role and h+ plays a minor role in the photocatalytic process. We believe that our findings can open a new avenue for the photocatalytic applications of Ag@MnO2 nanowires.
1. Introduction
Organic compounds are important pollutants in water and they come from various processes in industries such as food, textiles, leather, pharmaceutical and plastic.1,2 These organic pollutants are harmful to the environment, hazardous to human health and difficult to decompose which may be due to their semi volatility, low water solubility, high lipid solubility, persistence and high molecular weight. Therefore, many treatment approaches have been investigated for the removal of dyes from natural waters and wastewater streams including photochemical degradation, biological degradation, chemical oxidation, ultrasound degradation, coagulation and flocculation, advanced oxidation processes (AOPs) and adsorption.3–9 Among them, AOPs have been commonly suggested as a cost effective method for the complete mineralization of many toxic and non-biodegradable organic pollutants from aqueous systems.10–12In recent years, semiconductors based on metal oxides have been given much attention because of their high stabilities, non-toxicities and chemical properties. Several metal oxide semiconductors such as TiO2, ZnO, SnO2, WO3, NiO, CeO2 (ref. 13–18) etc., that are used widely for environmental remediation have a sufficient energy gap to catalyze photochemical reactions. Among these materials, manganese dioxide (MnO2) has been considered as a promising photocatalyst for oxidative degradation of organic and inorganic contaminants in wastewater due to its mass of merits like low-cost, non-toxic properties, ease of synthesis, acid resistance, strong adsorption and strong oxidation ability.19–22 Up to now, Zou et al. reported the higher photocatalytic degradation of the prepared MnO2/TiO2 nanocomposite for methylene blue under visible light radiation.23 MnO2 based photocatalysts have been reported by Ye et al.,24 Liu et al.,25 Ma et al.,26 Xu et al.,27 Yu et al.,28 Ramesh et al.29 and Bai et al.30 for the photocatalytic degradation of organic dyes. However, the efficiency of MnO2 as a photocatalyst is often restricted, because of the higher recombination rate of the photogenerated electron–hole pairs. Meanwhile, the morphology and structure also have important parameters on the rate of photocatalytic efficiency. In order to develop the photocatalyst material, the doping of noble metals such as Ag, Au, Pd, and Pt which could conduct electrons and delay/prevent the recombination efficiency of the photogenerated charges facilitates the improved photocatalytic activity.
Nowadays, doping of noble metal nanoparticles (Pd, Ag, Pt, Au) in semiconductors could improve the charge transfer by trapping the photo induced charge carriers and enhance the photochemical degradation process which may be due to the lower recombination of the electron–hole pairs and strong localized surface plasmon resonance.31–33 Compared with other noble metals, Ag could be a promising mediator for industrial applications due to its low cost, good conductivity and non-toxicity. Recent studies have reported the tremendous visible light response of Ag-based semiconductor materials including Ag–SnO2,34 Ag–ZnO,35 Ag–TiO2,36 Ag/CeO2,37 and Ag/ZrO2.38 These materials have an excellent photocatalytic activity because of the surface plasmon resonance effect (SPR) from the nanoparticles of metallic Ag under light irradiation. But the photocatalytic activities of the previously reported Ag doped composites showed a slightly poor activity, need an external oxidizing agent and are limited up to some dyes viz. rhodamine B, crystal violet, congo red and phenolic compounds.
Herein, for the first time, we report a novel Ag@MnO2 nanowires photocatalyst with an enhanced visible light response synthesized via the facile hydrothermal process. The photocatalytic activities of these Ag@MnO2 nanowires were investigated for the degradation of organic pollutants (Victoria Blue and Amaranth) under visible light irradiation. The enhanced photocatalytic performance of the Ag@MnO2 nanowires was attributed to their unique surface morphology and efficient separation of electron–hole pair recombination. In addition, the possible photocatalytic mechanism has been put forward based on the investigation of the reactive oxidative species. We believe that our findings can open up a new avenue for photocatalytic applications of Ag@MnO2 nanowires.
2. Materials and methods
2.1. Chemicals and materials
All of the chemical reagents were analytical grade and used as received without further purification. KMnO4, AgNO3, HCl and other chemicals were purchased from Merck, India. A commercial grade of Victoria Blue and Amaranth was used as a model organic pollutant. The structure and physicochemical properties of Victoria Blue and Amaranth are listed in Table 1. Deionized water was used for the preparation of all solutions.| Properties | Victoria Blue | Amaranth |
|---|---|---|
| Structure | 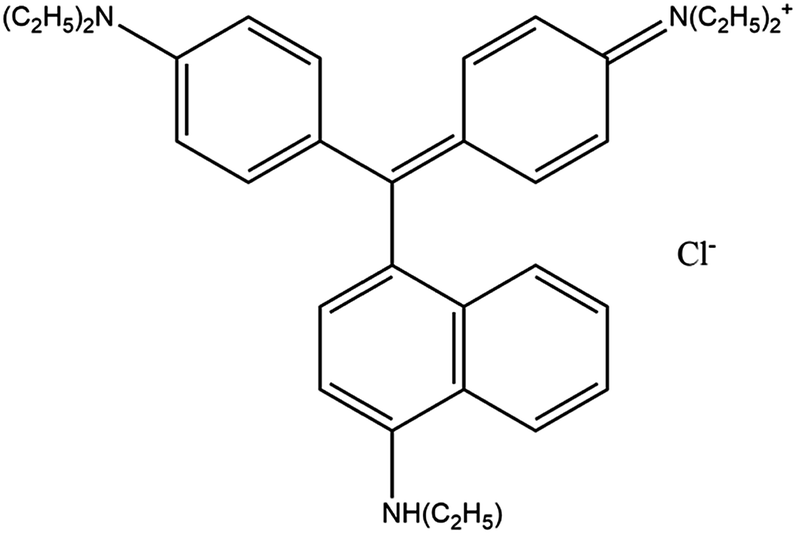 |
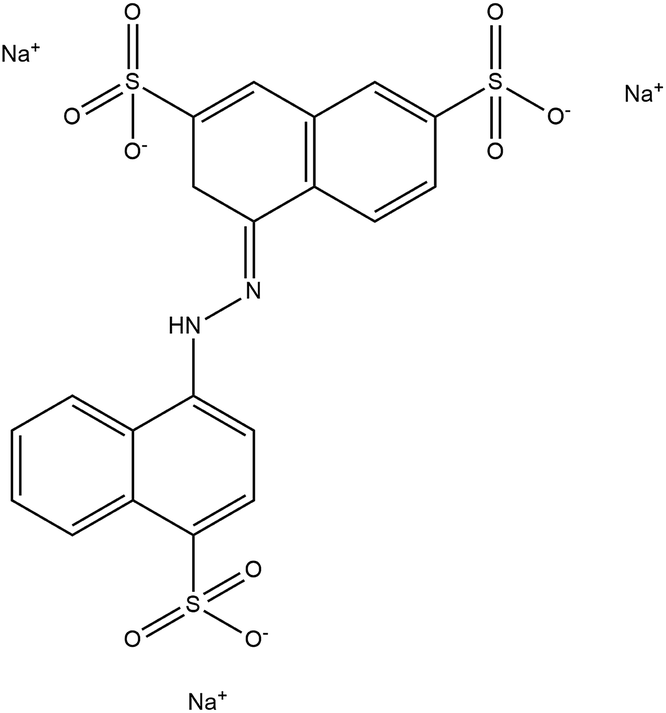 |
| Chemical formula | C33H40ClN3 | C20H11N2Na3O10S3 |
| Molecular weight | 514.14 g mol−1 | 604.48 g mol−1 |
| λmax | 615 nm | 521 nm |
| Stability | Soluble in water | Soluble in water |
2.2. Synthesis of the Ag@MnO2 photocatalyst
For the synthesis of the Ag@MnO2 catalyst, 2.83 g of KMnO4 was dissolved in 50 mL of deionized water using strong magnetic stirring. Then 5 mL of concentrated HCl solution was slowly dropped into the previous solution. Followed by 0.17 g of AgNO3 being added drop by drop into the above solution. The mixture of the solution was stirred continuously for 1 h to form a bluish black to light brown solution. Then the mixed solution was transferred into a 100 mL Teflon-lined stainless steel autoclave and heated in an oven at 180 °C for 12 h. After the autoclave was cooled down to room temperature in air, samples depositing at the bottom were collected and washed with DI water, and then dried at 60 °C for 12 h. Finally, Ag@MnO2 nanowires were obtained using annealing at 500 °C for 1 h. Pure MnO2 was synthesized through the same process as above without the addition of AgNO3 solution. The Ag@MnO2 nanowires synthetic process is schematically illustrated in Scheme 1.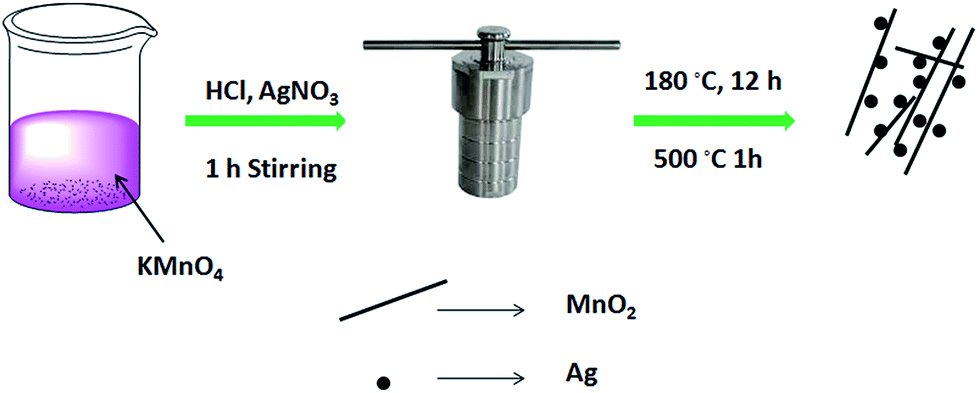 | ||
| Scheme 1 | ||
2.3. Characterization
The crystal phase and crystallite size of the samples were investigated using an {PAN alytical X’pert Pro} X-ray diffractometer measuring in the range 2θ of 10–80° with Cu Kα of 1.54178 Å. Morphologies and microstructures were studied using scanning electron microscopy {SEM, VEGA3 TESCAN} and transmission electron microscopy {TEM, PHILIPS CM 200 model}. Elemental analysis was examined using an EDX-Bruker Nano GmbH, X 50 Flash Detector (Model-5010). Fourier transform infrared spectroscopy measurements were carried out using a {Shimadzu FT-IR 3000 resolution 4 cm−1} spectrometer. The nitrogen adsorption–desorption isotherms were recorded at −196 °C using a micromeritics surface area analyzer model ASAP2020. The UV-vis diffuse reflectance spectra (DRS) of the sample were obtained on a UV-vis spectrophotometer {UV-2450, Shimadzu Corporation, Japan} using BaSO4 as the reference. The photoluminescence (PL) emission spectra of the as prepared samples were obtained using a {Hitachi F-7000} fluorescence spectrophotometer at room temperature. The electrochemical impedance spectra (EIS) characterization of the synthesized materials was carried out using an electrochemical workstation (CHI 660) with a three electrode system in a 2 M KOH solution. Photocurrent studies were measured using an electrochemical workstation (CHI660D) in a conventional three electrode system with a Pt wire as the counter electrode and Ag/AgCl as the reference electrode. A 100 W halogen lamp served as a visible light source. The absorption spectra in the photodegradation process were monitored with a {Shimadzu 2600} UV-vis spectrometer.2.4. Photocatalytic activity tests
The photocatalytic activities of the as-prepared samples were evaluated using the degradation of VB and Amaranth aqueous solution at room temperature under visible light irradiation. In the photocatalytic experiments, 50 mg of the catalysts was mixed with 100 mL of dye solution (10 mg L−1) in the dark and magnetically stirred for 30 min to ensure an absorption–desorption equilibrium between the photocatalyst and dye solution. A 500 W tungsten lamp equipped with a UV cut-off filter (λ > 400 nm) was used as the excitation source for the photocatalytic reaction. At varied irradiation time intervals, the change in the dye concentration was measured by monitoring the absorbance peak of Victoria Blue at λ = 615 and Amaranth at λ = 521 nm using a UV-vis spectrophotometer. All the photocatalytic measurements were carried out at room temperature. The photocatalytic degradation efficiency was calculated using C/C0, where C is the concentration of remaining organic dye solution at each time interval while C0 is the initial concentration of the dye solution. In addition, the stability of the catalyst was also studied by washing and drying the catalyst before the next run.To investigate the active species generated in the photocatalytic process, the trapping experiments of free radicals (hole radicals, hydroxyl radicals and superoxide radicals) capture were performed using ammonium oxalate, t-butyl alcohol and benzoquinone respectively.
3. Results and discussion
3.1. Structural characterization
X-ray diffraction (XRD) analysis, performed to confirm the crystal structures of the final products, is presented in Fig. 1. The XRD peaks at the 2θ values of 12.7°, 18°, 28.8°, 32.9°, 37.5°, 38.9°, 42.1°, 49.8°, 56.1°, 60.2°, 65.3° and 69.6° corresponding to the (110), (200), (310), (211), (330), (301), (411), (660), (521), (002) and (541) crystal faces of α-MnO2, matched well with the reported values from the Joint Committee on Powder Diffraction Standards card (JCPDS card no. 44-0141). The crystal phase of the α-MnO2 can be indexed as a highly crystalline nature and the tetragonal structure of α-MnO2. The average crystallite size of α-MnO2 can be calculated from the (211) peak of the XRD pattern using the Debye–Scherrer formula. Whereas in curves (b–e), apart from the α-MnO2, the intensity of the Ag peaks appears gradually when the Ag content increases from 1% to 15% (JCPDS card no. 01-1167), indicating the formation of the Ag@MnO2 nanocomposites. Moreover, the diffraction peaks at 28.1°, 33° and 45.3° correspond to the (220), (311), and (420) reflection planes of metallic Ag with a cubic structure. From these results, the diffraction peaks of the Ag and α-MnO2 phases were observed for all samples; and no other diffraction peaks were found.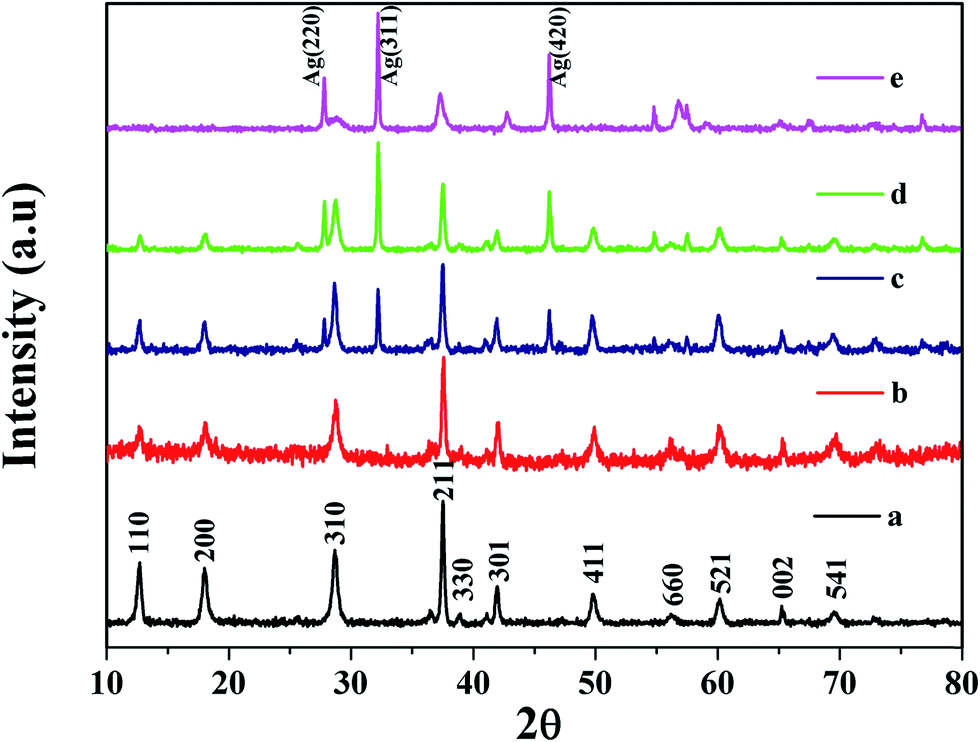 | ||
| Fig. 1 XRD patterns of the as-prepared (a) pure MnO2 and the Ag@MnO2 nanocomposites with different Ag content: (b) 1%, (c) 5%, (d) 10% and (e) 15%. | ||
The average crystalline size of the α-MnO2 and Ag@MnO2 nanowires was estimated using Scherrer’s equation as follows,39
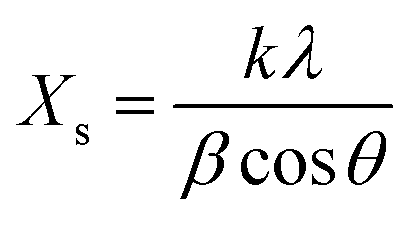 | (1) |
FTIR spectra of MnO2 and the Ag@MnO2 nanowires recorded in the region of 400–4000 cm−1 are shown in Fig. S1.† In the FTIR spectra analysis, the three main peaks observed at 470, 526 and 718 cm−1 could be attributed to the Mn–O stretching vibrations. The appearance of peaks at 1632 and 3425 cm−1 correspond to the fundamental bending vibration of the O–H mode and the stretching vibration of the surface hydroxyl groups, respectively.40 Thus, the FTIR results clearly verify the formation of the MnO2 and Ag@MnO2 nanowires and they were in good agreement with the XRD results. No other peaks were detected in the FTIR spectrum.
3.2. Surface area and porosity
The specific surface area and porous structure of the MnO2 and Ag@MnO2 nanowires were studied using nitrogen adsorption–desorption analysis which is critical to photocatalysts for enhanced photocatalytic activity. The N2 adsorption–desorption isotherms and the Barrett–Joyner–Halenda (BJH) pore size distribution (inset) are shown in Fig. 2. The Brunauer–Emmett–Teller (BET) specific surface area of the pure MnO2 nanowires was measured to be about 15.7 m2 g−1 from the N2 adsorption–desorption isotherm which is slightly higher than Ag@MnO2 (14.9 m2 g−1). The pore size distributions calculated from the desorption data using the (BJH) model indicate that the average pore diameter of MnO2 and Ag@MnO2 are 33.1 nm and 29.5 nm, respectively. The larger specific surface area can accommodate more surface active sites and facilitate the transport of charge carriers, which could lead to the enhanced photocatalytic performance.41,42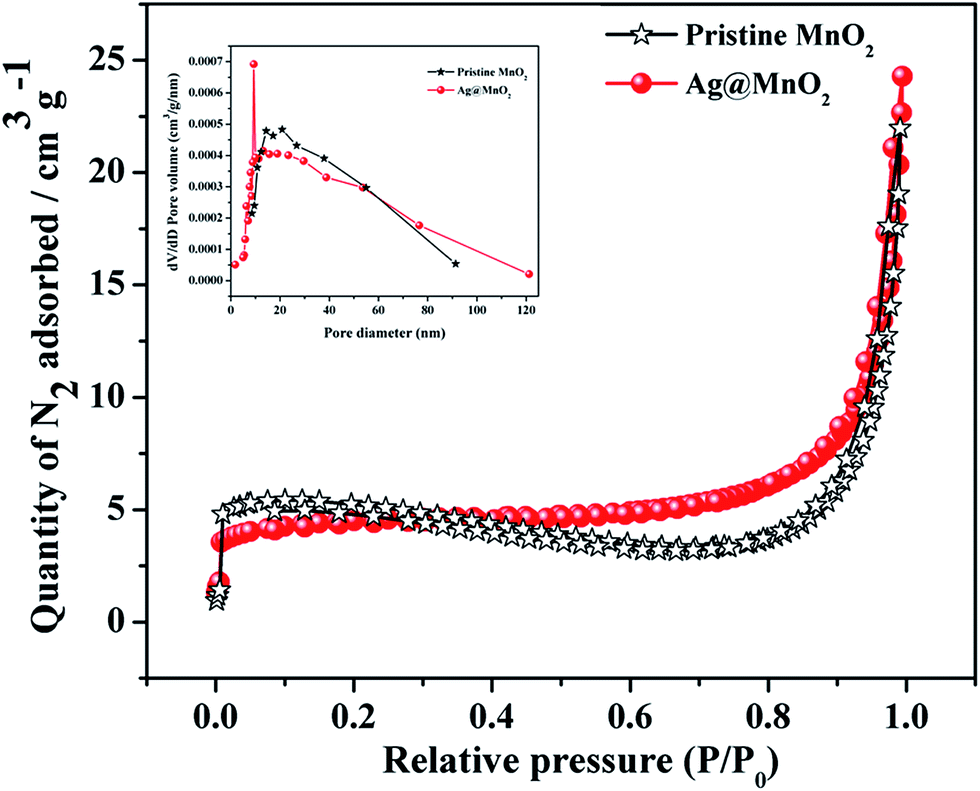 | ||
| Fig. 2 Nitrogen adsorption/desorption isotherms of pure MnO2 and Ag@MnO2 (5%). BJH pore size distribution curves are shown inset. | ||
3.3. Morphological characterization
The surface morphologies of the nanostructures of the MnO2 and Ag@MnO2 nanowires were observed using scanning electron microscopy (SEM) which is shown in Fig. 3. As shown in Fig. 3a and b, the SEM micrographs of the MnO2 nanowires confirmed the formation of entangled one-dimensional (1D) nanowires, with an average length of 2 to 5 μm and diameters varying from approximately 20 to 50 nm. As may be seen from the SEM micrographs, the nanowires were uniform in diameter through the entire length and were chaotically dispersed. After Ag was loaded on the MnO2 nanowires, the Ag nanoparticles (NPs) were highly anchored on the surfaces of the MnO2 nanowires which is shown in Fig. 3c and d. | ||
| Fig. 3 SEM micrographs of the (a, b) pure MnO2 and (c, d) Ag@MnO2 (5%) with different magnification. | ||
The detailed surface morphologies of the MnO2 and Ag@MnO2 nanowires were further evidenced using transmission electron microscopy (TEM). Fig. 4a and b display TEM images of MnO2, from which the 1D nanowire morphology can be clearly identified and the corresponding selected area diffraction (SAED) pattern (inset Fig. 4b) indicates that the MnO2 was of a single crystalline nature with tetragonal geometry. Fig. 4c and d showed that the Ag NPs anchored on the walls/surface of the MnO2 nanowires (marked with a circle in Fig. 4d) and the average particle size of the Ag NPs was 5–10 nm. As displayed in Fig. 4e, Ag@MnO2 exhibited the two distinct clear lattice fringes which showed d = 0.24 and d = 0.39 nm, corresponding to the (211) reflection plane of the MnO2 phase and the (311) plane of metallic Ag respectively. The results observed from the lattice fringes were in strong agreement with the XRD results. Besides, according to Fig. 4f, the image of the SAED pattern indicates a good crystallinity of the Ag@MnO2 nanowires. The SAED pattern further confirmed that the as prepared Ag@MnO2 nanowires, are well consistent with the XRD results.
 | ||
| Fig. 4 TEM images of (a, b) pure MnO2 inset shows the corresponding SAED pattern, (c, d) Ag@MnO2 (5%), (e) lattice fringes and (f) the corresponding SAED pattern. | ||
Moreover, the elemental composition of the as-prepared Ag@MnO2 nanowires was examined using energy dispersive spectroscopy (EDX). As expected, the spectrum displays the presence of three strong main peaks at around 0.5, 3 and 5.9 keV that correspond to the binding energies of O, Ag and Mn which is shown in Fig. S2.† EDX revealed the absence of any other element showing the purity of the as synthesized nanocomposites and the above findings confirmed that Ag is loaded on the MnO2 nanowires. The formation of the Ag@MnO2 nanowires was further evidenced using EDX elemental mappings and they are shown in Fig. S2.† The green, red and blue colors represent the distribution of silver, manganese and oxygen elements, respectively.
3.4. Optical properties
The desirable optical absorption and energy band gap were highly important for the photodegradation of dyes. Fig. 5a shows the UV-vis absorption (diffuse reflectance spectra mode) spectra of the pure MnO2 and Ag@MnO2 nanowires. From the spectra, it can be seen that the peak was absent in pure MnO2 and the absorption band edge enlarges to almost the full visible region.23 The Ag@MnO2 nanowires show a broad band in the visible light region, with an absorption edge of 350–600 nm. With the increase of Ag loading content, the absorption intensity of the Ag@MnO2 nanowires was enhanced gradually, which further confirms that Ag NPs had been successfully anchored on the surface of the MnO2 nanowires. The energy gap value was calculated using Tauc’s equation and the corresponding Tauc’s plot which is shown in Fig. 5b. The band gaps of the as-prepared pure MnO2 and Ag@MnO2 nanowires (1, 5, 10 and 15%) were 1.81 and 1.68, 1.56, 1.58 and 1.49 eV, respectively.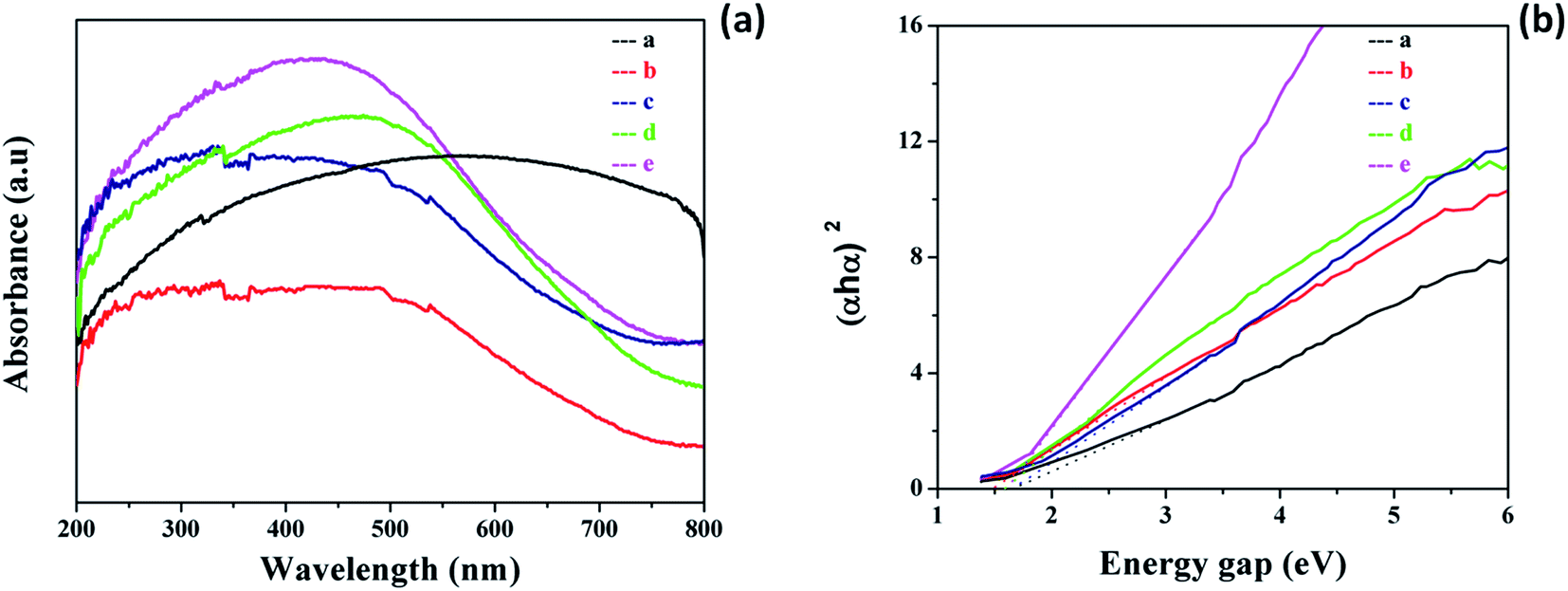 | ||
| Fig. 5 (a) UV-visible absorption spectra (DRS mode) of the pure MnO2 and Ag@MnO2 nanocomposites (with different Ag content), (b) Tauc’s plots of MnO2 and the Ag@MnO2 nanocomposites. | ||
The photoluminescence (PL) spectra were employed to characterize the charge transfer, photon-generated carrier trapping, migration and separation of the electron–hole pairs. In general, the lower the recombination rate of the electron–hole pairs the lower the PL intensity, which indicates the higher photocatalytic properties of the nanomaterials.43 Fig. 6 displays the PL spectra of the pure MnO2 and Ag@MnO2 nanowires with varying Ag content. In the spectra, the PL intensity of the Ag@MnO2 nanowires was smaller than that of pure MnO2, which indicates the recombination of the electron–holes is restrained effectively. The observed PL spectra exhibited three different emission peaks at 421, 460 and 484 nm in the visible region. The decrease in emission peak intensity for the Ag@MnO2 nanowires was due to the photo-generated electrons being able to transfer to Ag via an interface to accelerate the charge separation.44 The PL intensity decreased after the loading of Ag on the MnO2 which may be due to the Ag NPs on the MnO2 surface inhibiting the recombination of the electron–hole pairs. The PL intensity first decreases and then increases with the increase of Ag content which may be attributed to the aggregation of the Ag NPs.45 The 5% Ag@MnO2 nanowires showed the lowest PL emission intensity. Thus, it is clear that the PL study confirms the increased life time of the photogenerated carriers and subsequently favors the enhancement of photocatalytic performance. The separation efficiency of the photoexcited charge carriers plays a vital role in determining the photocatalytic performance of photocatalysts.
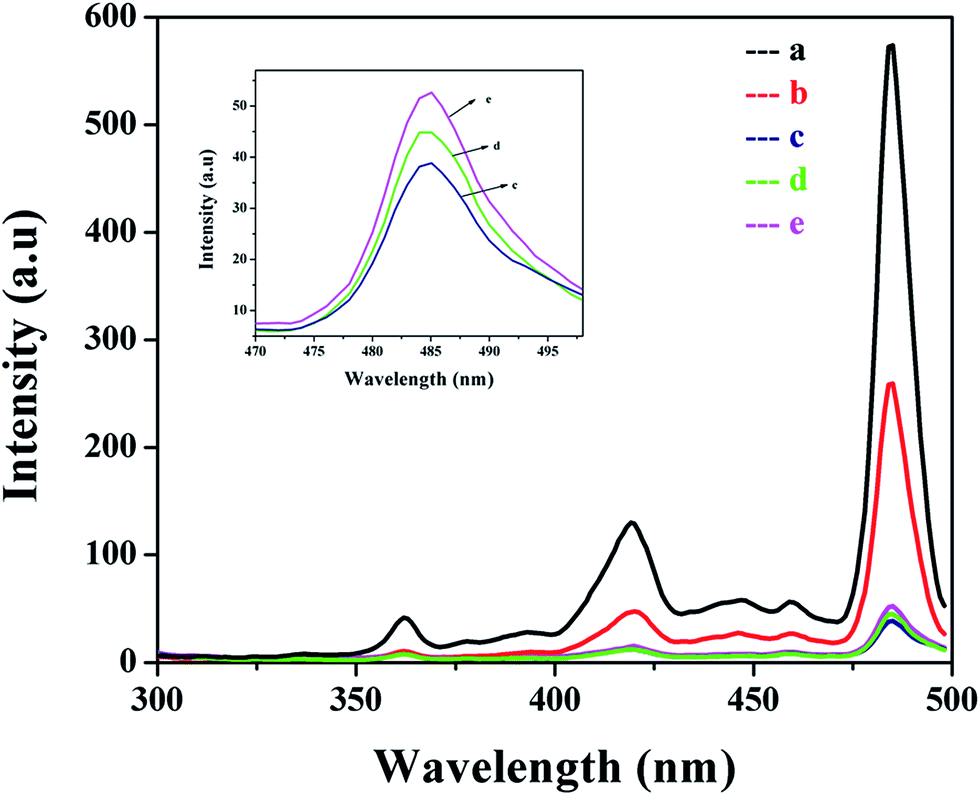 | ||
| Fig. 6 Photoluminescence spectra of (a) pure MnO2 and the Ag@MnO2 nanocomposites with different Ag content: (b) 1%, (c) 5%, (d) 10% and (e) 15%. Inset shows the large view of (c, d and e). | ||
3.5. Photoelectrochemical properties
The charge transfer resistance and recombination processes at the solid/electrolyte interfaces in the photocatalyst system can be studied using electrochemical impedance spectra (EIS). The charge separation efficiencies of the photogenerated electrons and holes is a critical factor for the photoactivity of photoelectrodes.46 The semicircle arc in the EIS spectra usually indicates the interface layer resistance of the surface of an electrode material. Fig. 7a shows the Nyquist plots of the MnO2 and Ag@MnO2 (5%) photocatalysts under visible light irradiation. Generally, a smaller arc radius indicates a higher charge transfer efficiency. It can be found that the arc radius of the Ag@MnO2 (5%) photocatalyst electrode is obviously smaller than the pure MnO2 photocatalyst, indicating a more effective separation of the photo-generated electron–hole pairs and a higher charge transfer rate occurred in the Ag@MnO2 (5%) photocatalyst which is greatly beneficial for the degradation of dye molecules.47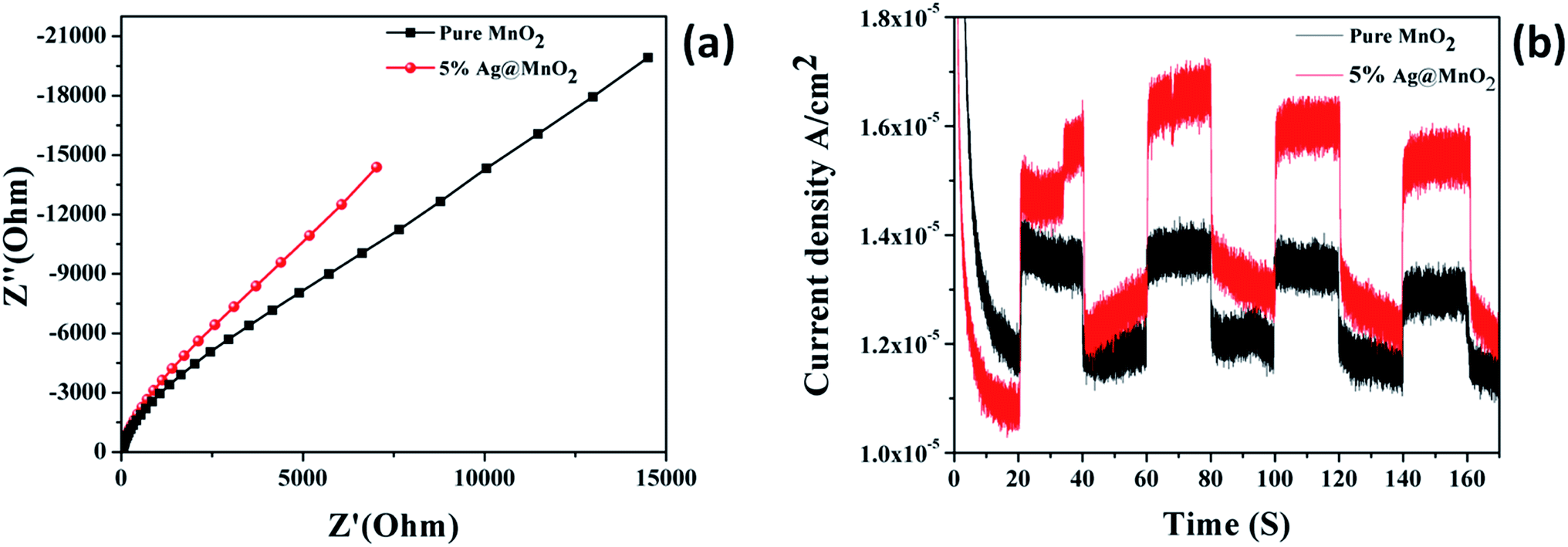 | ||
| Fig. 7 Electrochemical impedance spectra of pure MnO2 and Ag@MnO2 (5%) (a), transient photocurrent studies of pure MnO2 and Ag@MnO2 (5%) (b). | ||
Photocurrent studies were carried out to monitor the generation and transfer of the photoexcited charge carriers and also explain the interfacial charge transfer dynamics in the photocatalyst.48 Fig. 7b shows the curves of the photocurrent density values versus irradiation time over the MnO2 and Ag@MnO2 (5%) photocatalysts under visible light irradiation. Compared with the pure MnO2, the Ag@MnO2 (5%) exhibits an evidently enhanced photocurrent intensity under the same conditions, indicating the higher separation efficiency of the photoinduced electrons and holes. The photocurrent was created by the diffusion and separation of the photogenerated electrons and holes from the inner structure of the photocatalyst to the free charge acceptors on its surface.49 Thus, the effective decoration of Ag NPs on the MnO2 surface can enhance the light absorption abilities and facilitate effectively the separation efficiency of the charge carriers which might lead to enhanced photoactivity for the photodegradation of organic dyes.
3.6. Photocatalytic degradation
The photocatalytic activity of the as-prepared MnO2 and Ag@MnO2 nanowires was evaluated using VB and Amaranth dye degradation in the presence of visible light irradiation. As shown in Fig. 8 when the Ag@MnO2 nanowires were used as a photocatalyst, the degradation could be almost finished within 60 minutes of irradiation. The intensity of the peak continuously decreased with an increase in the irradiation time. In addition, the absence of the significant shift of absorbance during the reaction indicates that the Ag@MnO2 nanowires do not change the photodegradation pathway of the dye solutions in our system.50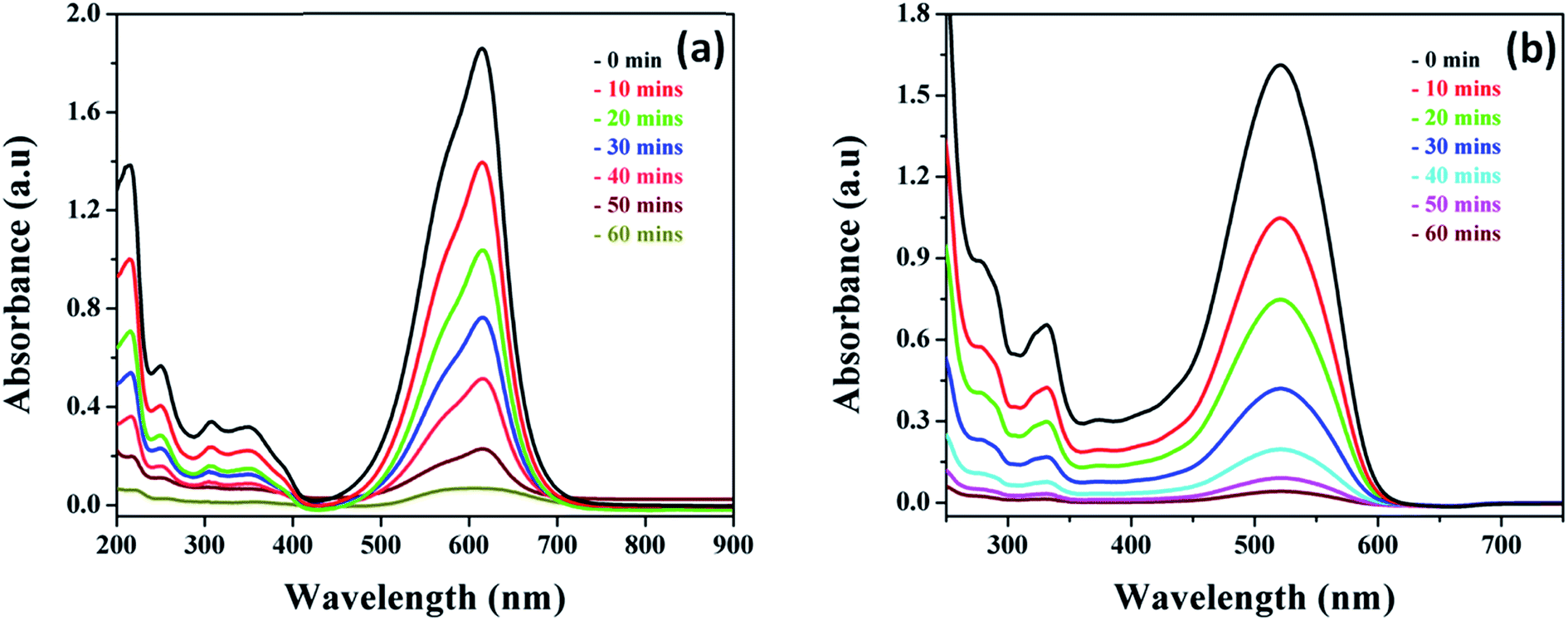 | ||
| Fig. 8 UV absorption spectra of different dye solutions (a) VB and (b) Amaranth (concentration = 10 mg L−1; catalyst dose = 50 mg; pH = 7) with Ag@MnO2 (5%) nanocomposite under visible light irradiation. | ||
As a comparison, VB dye was photodegraded using various loading amounts of Ag and this is shown in Fig. 9 and S3.† It was obvious that the photocatalytic activity of Ag@MnO2 was increased when the amount of Ag loading increased. However, beyond 5% Ag loading, a constant drop in the photocatalytic activity was observed which clearly indicated that over loading of Ag decreased the activity. The optimum loading amount of Ag was 5% and Ag@MnO2 (5%) shows an enhanced degradation rate of about 98%. Under visible light irradiation, Ag@MnO2 (5%) exhibited a superior photocatalytic performance compared to other Ag@MnO2 nanowires and pure MnO2, which can degrade VB within 60 min. The over loading of Ag content may create the aggregation effect, which will inhibit the physical contact between Ag and MnO2 that can decrease the photocatalytic activity. On the other hand, the separation efficiency of e−–h+ pairs decreased and further restrained the photocatalytic reaction which indicates the proper amount of Ag is an important factor for the photocatalytic process.51 Based on the above results, the photocatalytic activity of the as-prepared photocatalyst was in the order Ag@MnO2 (5%) > Ag@MnO2 (10%) > Ag@MnO2 (15%) > Ag@MnO2 (1%) > MnO2. In order to perform a blank experiment, the photolysis of a VB dye solution was carried out in the absence of the photocatalyst.
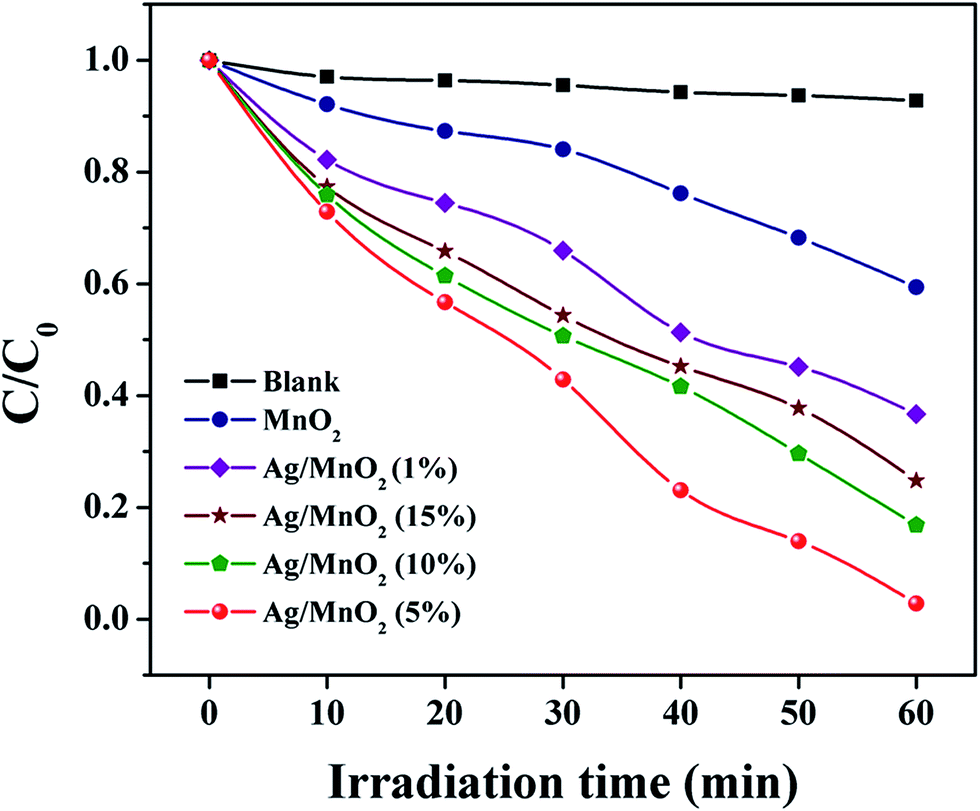 | ||
| Fig. 9 Absorption spectra of the VB dye solution under visible light in the presence of different catalysts. | ||
The optimum amount of photocatalyst is a crucial factor that controls the rate of photodegradation efficiency. The catalyst dose was varied from 10–75 mg and other parameters were kept the same (catalyst, dye concentration, pH and temperature) as shown in Fig. S4.† This observation showed that when the catalyst amount was increased from 10 mg, there was an increase in the rate of degradation for the photocatalytic reaction that preceded up to 50 mg. The maximum degradation efficiency was observed at 50 mg of catalyst and optimum catalyst dosage was found to be 50 mg of photocatalyst and this was used for the further photodegradation processes. Furthermore, over loading causes a negative impact on the added photocatalyst which will no longer affect the degradation rate which remains constant. After 50 mg of catalyst loading, the light penetration through the dye solution may be diminished and the unfavorable light scattering caused by the hindrance to the photons to reach the surface of the substrate.52
Furthermore, the photocatalytic degradation process strongly depends on the pH of the dye solutions. The effect of pH was also carried out in both an acidic and a basic medium for the photocatalytic degradation while the other conditions were unchanged which is shown in Fig. S5.† The initial pH of the dye solutions was adjusted using 0.1 M of HCl (acidic) and NaOH (basic). The degradation efficiency was found to be higher in the basic rather than the acidic medium, which may be due to the presence of more H+ ions in the acidic pH medium. The H+ ions could act as an electron scavenger and the H+ ions screen the active adsorption sites on the surface of the photocatalyst.53 The highest catalytic activity observed in the case of 5% of the Ag@MnO2 nanowires was 98% at pH 10. This is because, more OH− radicals were produced in the higher basic conditions which react with the photogenerated holes to form new active hydroxyl radicals.54
Fig. S6† shows the photocatalytic degradation of the dye solutions, carried out at various initial concentrations ranging from 10, 20 and 30 mg L−1. It was observed that the rate of degradation was inversely proportional to the initial concentration of dye. In this case, the rate of photocatalytic degradation efficiency was decreased with an increasing initial dye concentration which might be due to the reduction in the light intensity that reaches the active sites of the catalyst surface and the lower amount of ˙OH radical ions at a higher concentration of dye.55 In other words, the greater amount of dye molecules competing for the degradation process and the separation of the electron–hole pair was significantly reduced.
In order to reveal the photocatalytic oxidation process (POC), a series of reactive oxygen species such as h+, O2˙− and ˙OH play the bridging role in photocatalytic degradation under visible light irradiation.56 In the trapping experimental system, tert-butanol (t-BuOH) was used as an ˙OH radical scavenger, benzoquinone (BQ) was a scavenger of O2˙− and ammonium oxalate (AO) was used as a h+ scavenger. From Fig. 10 after the addition of t-BuOH and BQ, the photocatalytic degradation of VB was significantly prohibited, giving less than 50% VB degradation within 60 min of visible light illumination.57 On the contrary, no obvious effect on the photocatalytic activity was observed when AO was an added in the POC. This evidences that O2˙− and ˙OH radicals are key factors for VB photodegradation using the Ag@MnO2 nanowires under visible light.58,59
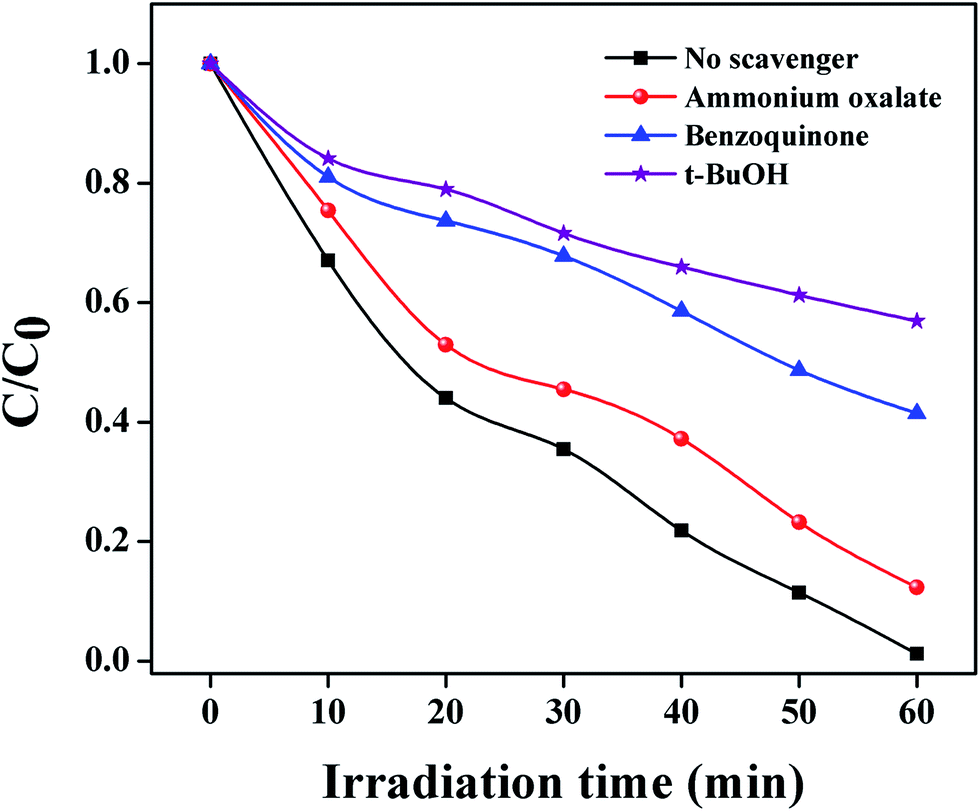 | ||
| Fig. 10 Impacts of different scavengers on the VB photodegradation under visible-light irradiation. | ||
The photocatalytic activity mechanism of the organic dyes in the presence of Ag@MnO2 can be summarized as follows,
| Ag@MnO2 + hν → Ag@MnO2 (e−) + (h+) | (2) |
| h+ + H2O → HO˙ | (3) |
| e− + O2 → O2˙− | (4) |
| O2˙− + H2O → HO2˙ + HO˙ | (5) |
| HO˙ + HO˙ → H2O2 | (6) |
| O2˙−/HO˙ + organic pollutant → mineralized product | (7) |
When the as-prepared catalyst is irradiated using visible light, the holes (h+) and electrons (e−) are generated in the valence band and conduction band of the photocatalyst simultaneously (eqn (2)). Then, the h+ will react with the hydroxyl groups or water molecules to produce hydroxyl radicals (eqn (3)). The conduction e− can be scavenged by the dissolved O2 on the surface of the catalyst to form superoxide radical anions (O2˙−) and further generate HO˙2 (eqn (4) and (5)). Hydrogen peroxide (H2O2) will be formed by the reaction between one HO˙ and another (eqn (6)). Finally, an organic pollutant could be largely oxidized by the photogenerated O2˙−, ˙OH and H2O2 radicals and generate a mineralized product like CO2, H2O etc. (eqn (7)).
The evaluation of the stability and reusability of the photocatalyst is vital for its practical applications. The photocatalytic measurement was performed with five consecutive cycles with illumination of visible light as shown in Fig. S7.† The experimental results showed that even after five cycles, the Ag@MnO2 nanowires were stable and the photocatalytic activity was slightly decreased. The XRD pattern shows no obvious crystalline phase structure change in the catalyst. However, a slight loss of photocatalytic activity was observed, probably due to some intermediates covered on the surface of the catalyst and photo dissolution of the catalyst.60 The obtained results revealed that the crystalline structure of the catalyst was well maintained even after five cycles which further confirms the good stability and reusable photocatalytic stability of the Ag@MnO2 nanowires.
4. Conclusion
In summary, a Ag NPs anchored on the surface of MnO2 nanowires photocatalyst was successfully synthesized through the simple hydrothermal method and the Ag@MnO2 (5%) nanowires showed enhanced photocatalytic activity compared to other Ag@MnO2 nanowires and pure MnO2. The significant enhancements in photocatalytic performance can be attributed to the efficient separation of the electron–hole pairs. The photoluminescence study indicated the reduced recombination rate which leads to the superior photodegradation efficiency under the visible light irradiation. Furthermore, photocatalytic mechanism investigations demonstrate that ˙OH and O2˙− play the key role in the photocatalytic process. The present study thus offers a facile and efficient synthesis protocol and a promising candidate catalyst for poisonous wastewater treatment in the near future.Acknowledgements
The authors acknowledge the support of the DST-SERB project New Delhi (ref no. SERB/F/4592/2013-14 dated 17.10.2013). We gratefully acknowledge the College managing board, Principal, and the Head of the department (Chemistry), VHNSN College for providing the necessary research facilities.References
- J. Ma, F. Yu, L. Zhou, L. Jin, M. X. Yang, J. S. Luan, Y. H. Tang, H. B. Fan, Z. W. Yuan and J. H. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 5749–5760 Search PubMed.
- E. Saputra, S. Muhammad, H. Sun, H. M. Ang, M. O. Tade and S. Wang, Appl. Catal., B, 2013, 142–143, 729–735 CrossRef CAS.
- L. Zhang, H. Li, Y. Liu, Z. Tian, B. Yang, Z. Sun and S. Yan, RSC Adv., 2014, 4, 48703–48711 RSC.
- J. Qu, L. Shi, C. He, F. Gao, B. Li, Q. Zhou, H. Hu, G. Shao, X. Wang and J. Qiu, Carbon, 2014, 66, 485–492 CrossRef CAS.
- Y. Liu, G. Cui, C. Luo, L. Zhang, Y. Guo and S. Yan, RSC Adv., 2014, 4, 55162–55172 RSC.
- N. Yuan, G. Zhang, S. Guo and Z. Wan, Ultrason. Sonochem., 2016, 28, 62–68 CrossRef CAS PubMed.
- S. W. Won, S. B. Choi, B. W. Chung, D. Park, J. M. Park and Y. S. Yun, Ind. Eng. Chem. Res., 2004, 43, 7865–7869 CrossRef CAS.
- W. Guo, X. Liu, P. Huo, X. Gao, D. Wu, Z. Lu and Y. Yan, Appl. Surf. Sci., 2012, 258, 6891–6896 CrossRef CAS.
- Y. Liu, C. Luo, J. Sun, H. Li, Z. Sun and S. Yan, J. Mater. Chem. A, 2015, 3, 5674–5682 RSC.
- R. Huang, Y. Liu, Z. Chen, D. Pan, Z. Li, M. Wu, C. H. Shek, C. M. Wu and J. K. Lai, ACS Appl. Mater. Interfaces, 2015, 7, 3949–3959 Search PubMed.
- K. T. Lee, X. F. Chuah, Y. C. Cheng and S. Y. Lu, J. Mater. Chem. A, 2015, 3, 18578–18585 RSC.
- Y. Su, Z. Wu, Y. Wu, J. Yu, L. Sun and C. Lin, J. Mater. Chem. A, 2015, 3, 8537–8544 RSC.
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- H. Qin, W. Li, Y. Xia and T. He, ACS Appl. Mater. Interfaces, 2011, 3, 3152–3156 Search PubMed.
- M. Alam and D. C. Cameron, Thin Solid Films, 2000, 377, 455–459 CrossRef.
- R. Abe, H. Takami, N. Murakami and B. Ohtani, J. Am. Chem. Soc., 2008, 130, 7780–7781 CrossRef CAS PubMed.
- L. Liu, Y. Li, S. M. Yuan, M. Ge, M. M. Ren, C. S. Sun and Z. Zhou, J. Phys. Chem. C, 2010, 114, 251–255 CrossRef CAS.
- J. Han, H. J. Kim, S. Yoon and H. Lee, J. Mol. Catal. A: Chem., 2011, 335, 82–88 CrossRef CAS.
- H. Zhao, G. Zhang and Q. Zhang, Ultrason. Sonochem., 2014, 21, 991–996 CrossRef CAS PubMed.
- W. F. Dong, L. H. Zang and H. Li, Appl. Mech. Mater., 2013, 361, 760–763 CrossRef.
- D. Zhao, X. Yang, H. Zhang, C. Chen and X. Wang, Chem. Eng. J., 2010, 164, 49–55 CrossRef CAS.
- M. Singh, D. N. Thanh, P. Ulbrich, N. Strnadova and F. Stepanek, J. Solid State Chem., 2010, 183, 2979–2986 CrossRef CAS.
- M. Xue, L. Huang, J. Q. Wang, Y. Wang, L. Gao, J. H. Zhu and Z. G. Zou, Nanotechnology, 2008, 19, 185604–185611 CrossRef PubMed.
- L. Ye, X. Liu, Q. Zhao, H. Xie and L. Zan, J. Mater. Chem. A, 2013, 1, 8978–8983 RSC.
- S. Liu, H. Liu, G. Jin and H. Yuan, RSC Adv., 2015, 5, 45646–45653 RSC.
- S. J. Li, Z. C. Ma, L. Wang and J. Z. Liu, Sci. China, Ser. B: Chem., 2008, 51, 179–185 CrossRef CAS.
- X. Xu, X. Zhou, X. Li, F. Yang, B. Jin, T. Xu, G. Li and M. Li, Mater. Res. Bull., 2014, 59, 32–36 CrossRef CAS.
- X. Yu, J. He, D. Wang, Y. Hu, H. Tian and Z. He, J. Phys. Chem. C, 2012, 116, 851–860 CrossRef CAS.
- M. Ramesh, H. S. Nagaraja, M. P. Rao, S. Anandan and N. M. Huang, Mater. Lett., 2016, 172, 85–89 CrossRef CAS.
- B. Bai, Q. Qiao, H. Arandiyan, J. Li and J. Hao, Environ. Sci. Technol., 2016, 50, 2635–2640 CrossRef CAS PubMed.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Phys. Chem. B, 2003, 107, 7479–7985 CrossRef CAS.
- M. J. Height, S. E. Pratsinis, O. Mekasuwandumrong and P. Praserthdam, Appl. Catal., B, 2006, 63, 305–312 CrossRef CAS.
- X. Liu, L. Pan, T. Lv, Z. Suna and C. Sun, RSC Adv., 2012, 2, 3823–3827 RSC.
- S. A. Ansari, M. M. Khan, M. O. Ansari, J. Lee and M. H. Cho, RSC Adv., 2014, 4, 26013–26021 RSC.
- S. A. Ansari, M. M. Khan, M. O. Ansari, J. Lee and M. H. Cho, J. Phys. Chem. C, 2013, 117, 27023–27030 CrossRef CAS.
- A. Shet and K. Vidya Shetty, Sol. Energy, 2016, 127, 67–78 CrossRef CAS.
- K. Saravanakumar, M. Mymoon Ramjan, P. Suresh and V. Muthuraj, J. Alloys Compd., 2016, 664, 149–160 CrossRef CAS.
- J. Zhang, L. Li, S. Wang, T. Huang, Y. Hao and Y. Qi, RSC Adv., 2016, 6, 13991–14001 RSC.
- P. S. Kumar, M. Selvakumar, S. G. Babu, S. K. Jaganathan, S. Karuthapandian and S. Chattopadhyay, RSC Adv., 2015, 4, 57493–57501 RSC.
- P. Muthukrishnan, K. Saravanakumar, B. Jeyaprabha and P. Prakash, Metall. Mater. Trans. A, 2014, 45, 4510–4524 CrossRef CAS.
- Y. Shi, Z. Yang, Y. Liu, J. Yu, F. Wang, J. Tong, B. Sua and Q. Wang, RSC Adv., 2016, 6, 39774–39783 RSC.
- K. Zhang, F. J. Zhang, M. L. Chen and W. C. Oh, Ultrason. Sonochem., 2011, 3, 765–772 CrossRef PubMed.
- P. Senthil Kumar, M. Selvakumar, S. Ganesh Babu, S. Kumar Jaganathan, S. Karuthapandian and S. Chattopadhyay, RSC Adv., 2015, 5, 57493–57501 RSC.
- J. P. Huo, L. T. Fang, Y. L. Lei, G. C. Zeng and H. P. Zeng, J. Mater. Chem. A, 2014, 2, 11040–11044 RSC.
- X. Z. Li and Y. Q. Wang, J. Alloys Compd., 2011, 509, 5765–5768 CrossRef CAS.
- M. M. Khan, S. A. Ansari, J. H. Lee, M. O. Ansari, J. Lee and M. H. Cho, J. Colloid Interface Sci., 2014, 431, 255–263 CrossRef CAS PubMed.
- S. Vadivel, A. Nirmalesh Naveen, V. P. Kamalakannan, P. Cao and N. Balasubramanian, Appl. Surf. Sci., 2015, 351, 635–645 CrossRef CAS.
- Y. Hong, C. Li, G. Zhang, Y. Meng, B. Yin, Y. Zhao and W. Shi, Chem. Eng. J., 2016, 299, 74–84 CrossRef CAS.
- X. J. Bai, L. Wang, Y. J. Wang, W. Q. Yao and Y. F. Zhu, Appl. Catal., B, 2014, 152–153, 262–270 CrossRef CAS.
- L. P. Zhu, G. H. Liao, N. C. Bing, L. L. Wang, Y. Yang and H. Y. Xie, CrystEngComm, 2010, 12, 3791–3796 RSC.
- W. Yu, X. Liu, H. Chu, G. Zhu, J. Li, J. Liu, L. Niu, Z. Sun and L. Pan, J. Mol. Catal. A: Chem., 2015, 407, 25–31 CrossRef CAS.
- S. K. Kansal, A. H. Ali and S. Kapoor, Desalination, 2010, 259, 147–155 CrossRef CAS.
- P. R. Chowdhury and K. G. Bhattacharyya, Dalton Trans., 2015, 44, 6809–6824 RSC.
- G. Nagaraju, T. N. Ravishankar, K. Manjunatha, S. Sarkar, H. Nagabhushana, R. Goncalves and R. J. Dupont, Mater. Lett., 2013, 109, 27–30 CrossRef CAS.
- T. N. Ravishankar, K. Manjunatha, T. Ramakrishnappa, G. Nagaraju, D. Kumar, S. Sarakar, B. S. Anandakumar, G. T. Chandrappa, V. Reddy and J. Dupont, Mater. Sci. Semicond. Process., 2014, 26, 7–17 CrossRef CAS.
- S. J. Liang, S. Y. Zhu, Y. Chen, W. M. Wu, X. C. Wang and L. Wu, J. Mater. Chem., 2012, 22, 2670–2678 RSC.
- S. G. Babu, R. Vinoth, B. Neppolian, D. D. Dionysiou and M. Ashokkumar, J. Hazard. Mater., 2015, 291, 83–92 CrossRef CAS PubMed.
- K. Saravanakumar, P. Senthil Kumar, J. Vinoth Kumar, S. Karuthapandian, R. Philip and V. Muthuraj, Energy and Environment Focus, 2016, 5, 50–57 CrossRef.
- E. Akbarzadeh, S. R. Setayesh and M. R. Gholami, RSC Adv., 2016, 6, 14909–14915 RSC.
- Z. Jiang and J. Xie, RSC Adv., 2016, 6, 3186–3197 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10444d |
| This journal is © The Royal Society of Chemistry 2016 |