
Design of recyclable TEMPO derivatives bearing an ionic liquid moiety and N,N-bidentate group for highly efficient Cu(I)-catalyzed conversion of alcohols into aldehydes and imines†
Bin Guoa,
Jiang-Yan Xuea,
Hong-Xi Li*a,
Da-Wei Tana and
Jian-Ping Lang*ab
aState and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou 215123, Jiangsu, P. R. China. E-mail: lihx@suda.edu.cn; jplang@suda.edu.cn; Fax: +86-512-65880328
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China
First published on 20th May 2016
Abstract
Four different types of TEMPO derivatives incorporated with an ionic liquid moiety and N,N-bidentate coordination group (IL–TEMPO-N,N) were prepared. The CuBr/IL–TEMPO-N,N system showed high catalytic activity toward the synthesis of aldehydes and imines via the aerobic oxidation of alcohols in 1-butyl-3-methylimidazolium tetrafluoroborate ([bmim]BF4). Both the Cu catalyst and IL–TEMPO-N,N co-catalyst in homogeneous catalytic systems could simultaneously be recovered from the products by extraction using Et2O. The remaining catalyst system in the ionic liquid phase could be reused for several cycles without obvious loss of catalytic activity. Protocols for highly efficient and recyclable aerobic oxidation of alcohols to aldehydes and imines were established.
Introduction
The selective oxidation of alcohols into their corresponding carbonyl compounds is one of the most important reactions in both the industrial synthesis of fine chemicals and laboratory research.1–3 These transformations are traditionally performed with stoichiometric amounts of inorganic oxidants (e.g. CrO3, KMnO4, MnO2, SeO2, Br2), which generate environmentally hazardous or toxic byproducts.4 Consequently, increasing efforts have been made to develop clean processes for the synthesis of carbonyl compounds. Many highly efficient catalysts such as Pd,5 Pt,6 Ru,7 Rh,8 Au,9 and Fe10 complexes have been developed for these conversions. From the practical and environmental points of view, there is a strong demand for the screening out efficient catalytic systems using inexpensive and environmentally benign metal catalysts and non-toxic air or O2 as the sole terminal oxidant.11 Since the fundamental studies by Semmelhack et al.,12 the homogeneous Cu catalytic systems bearing various organic ligands in combination with 2,2,6,6-tetramethylpiperidinooxy (TEMPO) and its derivatives have been developed as a highly efficient method for the aerobic oxidation of alcohols using O2 or air as the terminal oxidant.13 Despite the high activity achieved by these dual catalytic systems, the separation and recycling of the copper catalysts and TEMPO from the products remains a key issue. In recent decades, much attention has focused on the recyclability of heterogeneous14 or water-soluble Cu catalysts.15 Additionally, TEMPO derivatives could be immobilized on silicates,16 fluorous supports,17 soluble and insoluble polymers,18 magnetic nanoparticles,19 and ions/ionic liquids.20 In the presence of a co-oxidant, these metal-free functionalized TEMPO catalysts provided a convenient tool for the oxidation of alcohols working as recyclable reservoirs. However, little research concerns how to achieve simultaneous recoverability of both TEMPO derivatives and Cu catalysts.21 Toy and co-authors developed a multipolymer system for the oxidation of alcohols to recover both TEMPO and Cu(II) catalyst at the same time.22 Rodionov et al. developed a functional surfactant architecture incorporating TEMPO moieties, Cu-binding sites and sulfonate groups.23 Kitagawa et al. reported a novel porous coordination polymer (PCP) decorated with nitroxyl radicals could be an efficient and recyclable catalyst for the selective oxidation of a variety of alcohols to aldehydes or ketones.24 Very recently, we have developed some well-defined water-soluble copper coordination complexes combined with TEMPO for the aerobic oxidation of alcohols to aldehydes and the ammoxidation of alcohols to nitriles in aqueous system.15 Gree, Ragauskas et al. reported TEMPO–CuCl25 or acetamido–TEMPO/Cu(ClO4)2/DMAP (DMAP = 4-(dimethylamino)pyridine) in 1-butyl-3-methylimidazolium hexafluorophosphate ([bmim]PF6)26 catalyze the aerobic oxidation of primary and secondary alcohols. Although these water-soluble copper catalysts and ionic liquids showed a good cycle performance, the additional portion of TEMPO was required for next runs. TEMPO and its derivatives are rather expensive and, consequently, their separation and reuse after the aerobic oxidation reaction is highly desirable. In order to fulfil the recovery of TEMPO and copper catalyst simultaneously, we have designed a set of TEMPO derivatives (IL–TEMPO-N,N, 1a–1d) integrated with both ionic liquid moiety and N,N-bidentate coordination group (Scheme 1). In such multi-functional IL–TEMPO-N,N, the incorporating N,N-bidentate group may anchor copper ion to increase its catalytic activity while the attached TEMPO fragment is used as a co-catalyst, and the ionic liquid tag is crucial to the recyclability of the catalysts. In this context, we found that this resulting IL–TEMPO-N,N/CuBr homogeneous catalyst system showed remarkably high catalytic activity toward the synthesis of aldehyde and imine from a wide range of alcohols via the aerobic oxidation of alcohols in [bmim]BF4. Moreover, both Cu catalyst and TEMPO co-catalyst can be readily reused at least six cycles.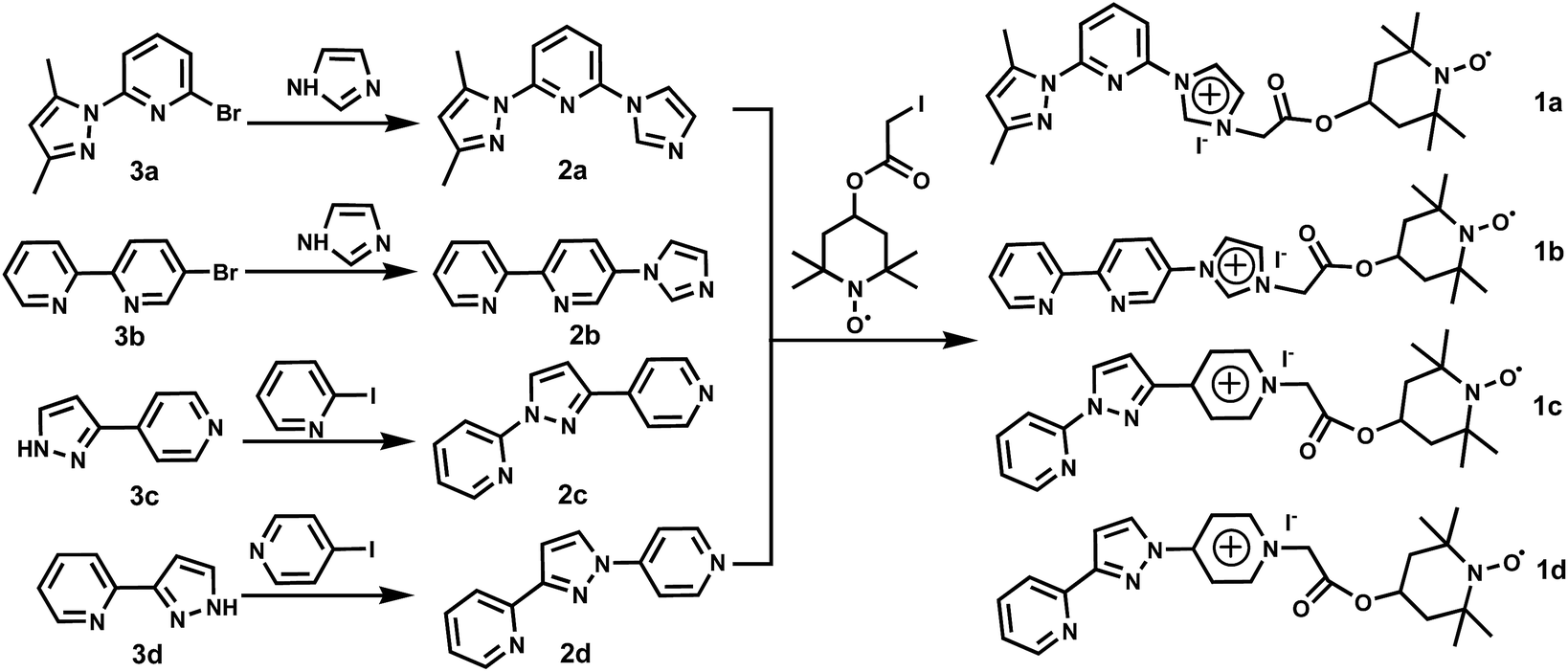 | ||
| Scheme 1 Synthesis of IL–TEMPO-N,N ligands 1a, 1b, 1c and 1d. | ||
Results and discussion
The multi-functional TEMPO derivatives reported in this work were obtained through a simple two-step synthesis starting from 2-bromo-6-(3,5-dimethyl-1H-pyrazol-1-yl)pyridine (3a), 5-bromo-2,2′-bipyridine (3b), 4-(1H-pyrazol-3-yl)pyridine (3c) and 2-(1H-pyrazol-3-yl)pyridine (3d), respectively. The synthesis routes of TEMPO radical derivatives bearing ion liquid and N,N-bidentate coordination groups (1a–1d) are depicted in Scheme 1. The first step involved the CuI-catalyzed cross-coupling reaction of 3a–3d with 1H-imidazole, 2-iodopyridine or 4-iodopyridine to give 2-(3,5-dimethyl-1H-pyrazol-1-yl)-6-(1H-imidazol-1-yl)pyridine (2a), 5-(1H-imidazol-1-yl)-2,2′-bipyridine (2b), 2-(3-(pyridin-4-yl)-1H-pyrazol-1-yl)pyridine (2c), 2-(1-(pyridin-4-yl)-1H-pyrazol-3-yl)pyridine (2d). Four IL–TEMPO-N,N derivatives (1a–1d) were prepared by the reactions of 2a, 2b, 2c, 2d with 4-(2-iodineacetoxy)-2,2,6,6-tetramethylpiperidine-1-oxyl free radical in high yields. 1a–1d are stable toward air and moisture, and freely soluble in H2O, MeOH, DMSO, CH2Cl2 and CHCl3, but insoluble in toluene and Et2O. Their IR spectra exhibit strong stretching vibrations at 1784 (1a), 1758 (1b), 1745 (1c) and 1753 (1d) cm−1, which are attributable to the carboxyl group. The high-resolution positive-ion mass spectra of 1a–1d revealed that there exists one peak at m/z = 452.2528 (1a), 435.2257 (1b), 435.2285 (1c) and 435.2282 (1d), which can be assigned to the [IL–TEMPO-N,N]+ ion. These TEMPO species are isolated as paramagnetic solids. Phenylhydrazine can reduce them into the corresponding IL–TEMPOH-N,N derivatives,20i which are identified by the in situ 1H NMR spectroscopy in CDCl3. 1H NMR spectra of IL–TEMPOH-N,N show the doublets at δ = 1.14 (1a), 1.13 (1b), 1.11 (1c) and 1.22 (1d) ppm, which are assigned as the protons of the methyl group. The multiplets at δ = 1.67/1.93/5.08 (1a), 1.65/1.93/5.07 (1b), 1.64/1.93/5.07 (1c) and 1.87/2.07/5.13 (1d) ppm are assigned as the protons of the TEMPO ring. The singlet at 5.46 (1a), 5.54 (1b), 5.82 (1c) or 5.93 (1d) ppm is ascribed to the protons of the methylene group.After these multi-functional IL–TEMPO-N,N derivatives (1a–1d) were obtained, 1a was chosen as a candidate to investigate its catalytic activity in combination with various copper salts using O2 as a terminal oxidant in an ionic liquid [bmim][BF4] (Table 1). At first, the oxidation reaction of p-tolylmethanol (4a) at 50 °C for 24 h produced 4-methylbenzaldehyde (5a) in 49% yield (Table 1, entry 1). The addition of K2CO3 (10 mol%) promoted the reaction to afford 5a in a higher yield (90%) (entry 2). To obtain optimized conditions for this reaction, we employed some other bases, such as Na2CO3, KOH, NaOH, Et3N and DMAP (entries 3–7). Na2CO3 proved to be the best one to give the highest yield (entry 3). The blank experiments revealed that no product was isolated in the absence of the copper catalyst or co-catalyst 1a (entries 8 and 9). Under the same conditions, CuBr/TEMPO catalytic system exhibited low activity (entry 10), which indicated the coordination group on 1a played an important role in its catalytic reactivity. Notably, the catalyst and base loading could be reduced from 10 to 5 mol% without affecting the product yields (entries 3, 11 and 12). When the reaction temperature was lowered from 50 °C to 45 °C to 35 °C, the yield of 5a was slightly decreased from 99% to 97% to 93% (entires 12, 14 and 15). Various Cu catalysts were screened, and CuCl, CuI, Cu(OAc)2 and Cu(NO3)2 exhibited slightly lower catalytic activity toward the oxidation of 4-methylbenzyl alcohol to 4-methylbenzaldehyde, as the desired product 5a was isolated in 42–86% yields (entries 16–19). To this end, the optimal reaction conditions were identified as follows: 5 mol% CuBr catalyst, 5 mol% Na2CO3 and 6 mol% 1a in [bmim][BF4] at 50 °C for 24 h. With these conditions, the catalytic performances of 1b–1d were also investigated. As a result, 1b–1d all worked well. The high activity of CuBr/1a–1d can be most likely attributed to the combination of ionic liquid moiety and N,N-bidentate coordination group.
|
||||
|---|---|---|---|---|
| Entrya | Cat. (mol%) | Ligand (mol%) | Base (mol%) | Yieldb (%) |
| a Reaction conditions: 4-methylbenzyl alcohol (1 mmol), [Cu], ligand, base, [bmim][BF4] (3 mL), O2 (1 atm), at 50 °C for 24 h.b GC yield.c 35 °C.d 45 °C. | ||||
| 1 | CuBr (10) | 1a (12) | — | 49 |
| 2 | CuBr (10) | 1a (12) | K2CO3 (10) | 90 |
| 3 | CuBr (10) | 1a (12) | Na2CO3 (10) | >99 |
| 4 | CuBr (10) | 1a (12) | DMAP (10) | 96 |
| 5 | CuBr (10) | 1a (12) | KOH(10) | 93 |
| 6 | CuBr (10) | 1a (12) | NaOH (10) | 94 |
| 7 | CuBr (10) | 1a (12) | Et3N (10) | 92 |
| 8 | — | 1a (12) | Na2CO3 (10) | Trace |
| 9 | CuBr (10) | — | Na2CO3 (10) | Trace |
| 10 | CuBr (10) | TEMPO (12) | Na2CO3 (10) | 65 |
| 11 | CuBr (5) | 1a (6) | Na2CO3 (10) | >99 |
| 12 | CuBr (5) | 1a (6) | Na2CO3 (5) | >99 |
| 13 | CuBr (3) | 1a (3.6) | Na2CO3 (5) | 43 |
| 14 | CuBr (5) | 1a (6) | Na2CO3 (5) | 93c |
| 15 | CuBr (5) | 1a (6) | Na2CO3 (5) | 97d |
| 16 | CuCl (5) | 1a (6) | Na2CO3 (5) | 42 |
| 17 | CuI (5) | 1a (6) | Na2CO3 (5) | 53 |
| 18 | Cu(OAc)2 (5) | 1a (6) | Na2CO3 (5) | 86 |
| 19 | Cu(NO3)2 (5) | 1a (6) | Na2CO3 (5) | 56 |
| 20 | CuBr (5) | 1b (6) | Na2CO3 (5) | 95 |
| 21 | CuBr (5) | 1c (6) | Na2CO3 (5) | 99 |
| 22 | CuBr (5) | 1d (6) | Na2CO3 (5) | 70 |

The scope of the aerobic oxidative reactions of alcohols was summarized in Table 2. Under the optimized conditions, a wide range of alcohols were smoothly oxidized into the corresponding aldehydes in good to excellent yields. As compared with bare benzyl alcohol, aromatic alcohols with substituted electron donating groups like –Me, –CMe3, –OMe and –OCH2O– exhibited a slightly increased conversion ratio (5a–5i, 92–99%). While the benzyl alcohols with electron-withdrawing substituents like –NO2, –Cl, –Br and –CF3 showed a decreased conversion ratio and required higher temperature (70 °C) to complete these transformations (5j–5m). The steric hindrance of benzyl alcohols also exerted impact on the catalytic activity of the transformation from benzylic alcohols to the corresponding benzaldehyde derivatives. The aromatic alcohols with a –Me or –OMe group substituted at different positions of phenyl ring showed different reaction rates in the order of ortho- < meta- < para-substituted (5b < 5c < 5a; 5d < 5e < 5f). (2,4,6-Trimethyl-phenyl)methanol was converted smoothly to the corresponding product in 88% yield (5t). The heteroatom-containing alcohols such as pyridin-3-ylmethanol, furan-2-ylmethanol and thiophen-2-ylmethanol underwent the aerobic oxidation reaction to produce the desired aldehydes (5n, 5o, 5p) in relatively high yields. Intriguingly, naphthalen-1-ylmethanol, 1,4-phenylenedimethanol and cinnamic alcohol could also be oxidized with satisfied yields under the standard reaction conditions (5q, 5r, 5s). We also examined the catalytic reactivity towards secondary alcohol. The oxidation of 1-phenylethan-1-ol under the similar reaction conditions gave the desired product acetophenone in 31% yield at 70 °C.


In contrast to the unmodified TEMPO, the catalytic species can be also separated by simple extraction of products and reused without further purification. The oxidation reaction of p-tolylmethanol was performed with CuBr (5 mol%), 1a (6 mol%) and Na2CO3 (5 mol%) in [bmim]BF4 to examine its reusability. After the first reaction run, the mixture was allowed to cool to room temperature for extraction with Et2O. The upper layer containing product was removed by decantation. The lower layer left was dried to remove a small amount of water by vacuum and fresh substrates were then recharged for the next run. As showed in Fig. 1, the catalyst system still went smoothly and the conversion got decreased slightly during four cycles, from 98% to 85%. At the fifth or sixth reuse run, a relatively high yield (84%, 79%) was still obtained after 36 h. These results indicated that the use of this catalytic system might meet the goal of green chemistry. To further demonstrate that the catalytic system is efficient and practicable, a scale-up reaction was carried out under the optimal conditions. The oxidation reaction of p-tolylmethanol (1.22 g, 10 mmol) gave the product in 78% isolated yield.
 | ||
| Fig. 1 The reusability of the CuBr/1a catalytic system for the aerobic oxidation of p-tolylmethanol into 4-methylbenzaldehyde. | ||
Table 3 lists a comparison of the results for the oxidation of phenylmethanol using different catalysts. Comparative runs with MPEG–Bipy/MPEG–TEMPO/CuBr2 (85%), CuCl/TEMPO–IL (85%) and [Cu(μ-Cl)(Cl)(phen)]2/TEMPO (83%) indicated that CuBr/1a exhibited higher catalytic activity (90%). At higher temperature, [Imim–PEG1000–TEMPO][CuCl2] and Cu–NHC–TEMPO showed higher activity than that of CuBr/1a.
| Entry | Cat. | Cat. loading (mol%) | Addition | Solvent | Temp (°C) | Yield (%) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | [Imim–PEG1000–TEMPO][CuCl2] | 5 | — | — | 60 | 96 | 21 |
| 2 | Cu–NHC–TEMPO | 10 | — | C6H5Cl | 80 | 96 | 20f |
| 3 | CuCl/TEMPO–IL | 10 | Molecular sieve 3A | [bmim][PF6] | 80 | 85 | 20l |
| 4 | MPEG–Bipy/MPEG–TEMPO/CuBr2 | 5 | t-BuOK | MeCN/H2O | 80 | 85 | 22 |
| 5 | Cu/TEMPO/amphiphile | 2 | DMAP | H2O | 25 | 94 | 23 |
| 6 | [Cu(μ-Cl)(Cl)(phen)]2/TEMPO | 5 | K3PO4 | MeCN | R.T. | 83 | 13a |
| 7 | Acetamido–TEMPO/Cu(ClO4)2 | 5 | DMAP | [bmim][PF6] | R.T. | 92 | 26 |
| 8 | Acetamido–TEMPO/Cu(ClO4)2/DMAP | 4 | DABCO | DMSO | R.T. | 98 | 13e |
| 9 | TEMPO/CuCl | 5 | — | [bmim][PF6] | 65 | 98 | 25 |
| 10 | [{Cu(NO3)}(μ-pzpypz)]2/TEMPO | 5 | K2CO3 | H2O | 30 | 95 | 15a |
| 11 | CuBr/1a | 5 | Na2CO3 | [bmim][BF4] | 50 | 90 | This work |
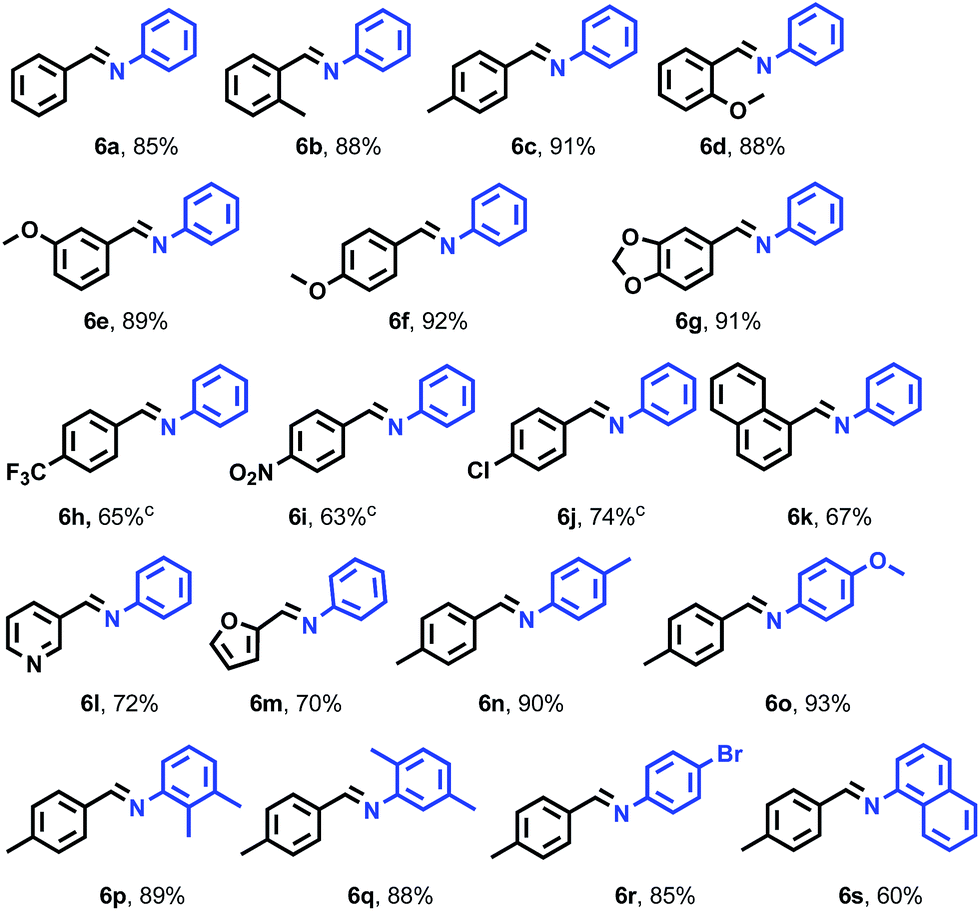
To explore more applications of the CuBr/1a catalytic system in organic synthesis, we then studied the aerobic oxidative coupling of benzyl alcohols with amines (Table 4). We also investigated the efficiency of one-pot cascade reaction between benzyl alcohol and amine to prepare imine at 80 °C in the presence of 5 mol% of CuBr and 6 mol% of 1a. After completion of this reaction (24 h), the reactions went well to give the desired imines in good to excellent yields (60–92%). The cascade reactions of aniline with benzyl alcohol derivatives with electron-donating substituents on the phenyl ring could afford smoothly the corresponding imines in high yields (6b–g). Benzyl alcohols with –Cl, –NO2 and –CF3 could also proceed smoothly at higher temperature (100 °C), but in lower yields (6h–j). The heteroaromatic benzyl alcohols also went on in moderate yields (6l and 6m). With respect to the aniline-derived moiety of the amine, the presence or absence of an electron-donating group did not significantly affect the yields of imines. Over 85% isolated yields were obtained uniformly (6n–s). For naphthalen-1-ylmethanol or naphthalen-1-amine, the corresponding imines were attained in 67% and 60% isolated yield (6k and 6s).

Conclusions
In summary, we showed that both ionic liquid and N,N-bidentate coordination groups were used to support TEMPO nitroxyl radical simultaneously, yielding multi-functional TEMPO derivatives IL–TEMPO-N,N. The IL–TEMPO-N,N/CuBr was found to be a highly efficient catalyst system for the formation of aldehyde and imine from alcohols in [bmim]BF4. Moreover, it could be recycled and reused without significant decay of catalytic activity after multiple runs. For this catalytic system, it would find a wider application in various reactions, which is ongoing in our laboratory.Experimental
General
All reagents were used as purchased from commercial sources without further purification. 2-(3,5-Dimethyl-1H-pyrazol-1-yl)-6-(1H-imidazol-1-yl)pyridine,27 2-(3-(pyridin-4-yl)-1H-pyrazol-1-yl)pyridine,28 and 2-(1-(pyridin-4-yl)-1H-pyrazol-3-yl)pyridine28 were prepared according to the published procedures. Column chromatography was performed on silica gel. 1H NMR and 13C NMR spectra were recorded at ambient temperature on a Varian UNITY plus-400 spectrometer.Synthesis of 5-(1H-imidazol-1-yl)-2,2′-bipyridine
A mixture of 5-bromo-2,2′-bipyridine (1.54 g, 6.6 mmol), 1H-imidazole (1.34 g, 19.7 mmol) and K2CO3 (1.81 g, 13.1 mmol) was added into a Schlenk tube equipped with a stirring bar. It was stirred at 190 °C for 18 h under a nitrogen atmosphere. After cooling to room temperature, the mixture was diluted in water and dichloromethane. The organic layer was separated, and the aqueous layer was extracted with dichloromethane for three times and washed three times with saturated aqueous Na2CO3 solution. The combined organic phases were dried over anhydrous Na2SO4 and filtrated. The solvent of the organic phase was removed under reduced pressure. Then, recrystallization of the orange residue from ethyl acetate (15 mL) afforded the white solid of 5-(1H-imidazol-1-yl)-2,2′-bipyridine (1.16 g, 80%). Mp 168.2–169.1 °C. HRMS calcd for C13H10N4: 222.0905; found: 223.0979 [M + H]+. Anal. calcd for C13H10N4: C, 70.26; H, 4.54; N, 25.21%. Found: C, 70.40; H, 4.73; N, 24.87%. 1H NMR (400 MHz, DMSO-d6): δ 9.06 (s, 1H), 8.69 (d, J = 3.8 Hz, 1H), 8.52–8.42 (m, 2H), 8.38 (d, J = 7.9 Hz, 1H), 8.25 (d, J = 8.5 Hz, 1H), 7.94 (dd, J = 15.5, 7.6 Hz, 2H), 7.52–7.39 (m, 1H), 7.19 (s, 1H). 13C NMR (101 MHz, DMSO-d6): δ 154.3, 153.4, 149.4, 141.1, 137.5, 135.7, 133.6, 130.4, 128.7, 124.3, 121.1, 120.4, 117.9.Synthesis of 2,2,6,6-tetramethylpiperidinooxy-4-yl 2-iodoacetate
This compound was prepared in a similar manner to chloroacetic acid 2,2,6,6-tetramethyl-1-oxy-piperidin-4-yl ester.20i A mixture of 2-iodoacetic acid (0.78 g, 4.2 mmol), dicyclohexylcarbodiimide (0.95 g, 4.6 mmol), and 4-dimethyl-aminopyridine (0.05 g, 0.4 mmol) was added into an anhydrous CH2Cl2 solution (15 mL) of 4-hydroxy-2,2,6,6-tetramethylpiperdine-1-oxyl (0.72 g, 4.2 mmol). The resulting mixture were stirred at room temperature and checked by TLC. After the substrate was consumed, 50 mL H2O was added. The mixture was extracted with CH2Cl2 for three times. The combined organic layer was dried over anhydrous Na2SO4 and concentrated under vacuum to obtain a crude product, which was further purified by flash chromatography (petroleum ether–ethylacetate, v/v = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to afford the red solid of 2,2,6,6-tetramethylpiperidinooxy-4-yl 2-iodoacetate (0.85 g, 59%). HRMS calcd for C11H19INO3: 340.0410; found: 341.0463 [M + H]+. Anal. calcd for C11H19INO3: C, 38.84; H, 5.63; N, 4.12%. Found: C, 39.31; H, 5.65; N, 4.27%. 1H NMR (400 MHz, CDCl3): δ 4.98 (ddd, J = 15.5, 11.2, 4.2 Hz, 1H), 3.54 (s, 2H), 1.88–1.79 (m, 2H), 1.55 (t, J = 11.8 Hz, 2H), 1.13 (d, J = 12.5 Hz, 12H). IR (KBr disk, cm−1): 3046 (w), 2965 (w), 1715 (s), 1466 (w), 1416 (w), 1311 (m), 1281 (s), 1178 (m), 1093 (s), 984 (m).
:1) to afford the red solid of 2,2,6,6-tetramethylpiperidinooxy-4-yl 2-iodoacetate (0.85 g, 59%). HRMS calcd for C11H19INO3: 340.0410; found: 341.0463 [M + H]+. Anal. calcd for C11H19INO3: C, 38.84; H, 5.63; N, 4.12%. Found: C, 39.31; H, 5.65; N, 4.27%. 1H NMR (400 MHz, CDCl3): δ 4.98 (ddd, J = 15.5, 11.2, 4.2 Hz, 1H), 3.54 (s, 2H), 1.88–1.79 (m, 2H), 1.55 (t, J = 11.8 Hz, 2H), 1.13 (d, J = 12.5 Hz, 12H). IR (KBr disk, cm−1): 3046 (w), 2965 (w), 1715 (s), 1466 (w), 1416 (w), 1311 (m), 1281 (s), 1178 (m), 1093 (s), 984 (m).
Synthesis of 3-(6-(3,5-dimethyl-1H-pyrazol-1-yl)pyridin-2-yl)-1-(2-((1-oxyl-2,2,6,6-tetramethylpiperidin-4-yl)oxy)-2-oxoethyl)-1H-imidazol-3-ium iodide (1a)
A solution of 2,2,6,6-tetramethylpiperidinooxy-4-yl 2-iodoacetate (0.66 g, 1.9 mmol) in anhydrous THF (10 mL) was treated with a solution of 2-(3,5-dimethyl-1H-pyrazol-1-yl)-6-(1H-imidazol-1-yl)pyridine (0.39 g, 1.6 mmol) in anhydrous THF (10 mL). The mixture was stirred at 50 °C for 12 h under N2 atmosphere. During this time, a creamcolored solid precipitated in the flask. The suspension was cooled to room temperature, and the supernatant was decanted off. The resulting solid was washed with anhydrous THF and anhydrous ether for three times to afford the creamcolored solid of 1a (0.60 g, 81%). HRMS calcd for C24H32N6O3+: 452.2530; found: 452.2528. Anal. calcd for C24H32N6O3I: C, 49.75; H, 5.57; N, 14.50%. Found: C, 49.61; H, 5.57; N, 14.24%. 1H NMR (400 MHz, CDCl3): δ 10.83 (s, 1H), 7.97 (d, J = 8.1 Hz, 1H), 7.89 (d, J = 7.9 Hz, 1H), 7.85 (d, J = 7.1 Hz, 1H), 7.80 (d, J = 7.8 Hz, 1H), 7.46 (s, 1H), 5.96 (s, 1H), 5.46 (s, 2H), 5.12–5.04 (m, 1H), 2.59 (s, 3H), 2.20 (s, 3H), 1.95–1.90 (m, 2H), 1.67 (t, J = 11.8 Hz, 2H), 1.14 (d, J = 23.2 Hz, 12H). IR (KBr disk, cm−1): 3084 (w), 2976 (w), 1784 (s), 1612 (m), 1586 (m), 1484 (m), 1453 (s), 1384 (w), 1360 (m), 1280 (w), 1220 (s), 811 (m), 725 (w).Synthesis of 3-([2,2′-bipyridin]-5-yl)-1-(2-((1-oxyl-2,2,6,6-tetramethylpiperidin-4-yl)oxy)-2-oxoethyl)-1H-imidazol-3-ium iodide (1b)
Creamcolored solid of 1b was obtained by the similar approach to that used for the isolation of 1a, using 2,2,6,6-tetramethylpiperidinooxy-4-yl 2-iodoacetate (0.57 g, 1.7 mmol) and 5-(1H-imidazol-1-yl)-2,2′-bipyridin (0.31 g, 1.4 mmol) as starting materials. Yield: 0.45 g (74%). HRMS calcd for C24H29N5O3+: 435.2265; found: 435.2257. Anal. calcd for C24H29N5O3I: C, 51.25; H, 5.20; N, 12.45%. Found: C, 50.60; H, 5.29; N, 12.41%. 1H NMR (400 MHz, CDCl3): δ 10.20 (s, 1H), 8.84 (s, 1H), 8.59 (d, J = 4.1 Hz, 1H), 8.51 (d, J = 8.6 Hz, 1H), 8.30 (d, J = 7.8 Hz, 1H), 8.09 (d, J = 8.6 Hz, 1H), 7.73 (t, J = 7.4 Hz, 1H), 7.57 (s, 1H), 7.41 (s, 1H), 5.45 (s, 2H), 5.11–5.03 (m, 1H), 1.94–1.89 (m, 2H), 1.65 (t, J = 11.9 Hz, 2H), 1.13 (d, J = 21.5 Hz, 12H). IR (KBr disk, cm−1): 2974 (s), 2936 (m), 2911 (m), 1758 (s), 1625 (w), 1563 (m), 1459 (s), 1360 (m), 1216 (s), 800 (m), 740 (m).Synthesis of 1-(2-((1-oxyl-2,2,6,6-tetramethylpiperidin-4-yl)oxy)-2-oxoethyl)-4-(1-(pyridin-2-yl)-1H-pyrazol-3-yl)pyridin-1-ium iodide (1c)
Creamcolored solid of 1c was obtained by the similar route to that used for the isolation of 1a, using 2,2,6,6-tetramethylpiperidinooxy-4-yl 2-iodoacetate (0.74 g, 2.2 mmol) and 2-(3-(pyridin-4-yl)-1H-pyrazol-1-yl)pyridine (0.40 g, 1.8 mmol) as starting materials. Yield: 0.75 g (74%). HRMS calcd for C24H29N5O3+: 435.2265; found: 435.2285. Anal. calcd for C24H29N5O3I: C, 51.25; H, 5.20; N, 12.45%. Found: C, 51.04; H, 5.33; N, 12.06%. 1H NMR (400 MHz, CDCl3): δ 8.91 (d, J = 6.4 Hz, 2H), 8.56 (d, J = 2.2 Hz, 1H), 8.33 (d, J = 4.1 Hz, 1H), 8.17 (d, J = 6.4 Hz, 2H), 7.94 (d, J = 8.2 Hz, 1H), 7.77 (t, J = 7.6 Hz, 1H), 6.92 (d, J = 2.2 Hz, 2H), 5.82 (s, 2H), 5.14–5.00 (m, 1H), 1.96–1.90 (m, 2H), 1.64 (t, J = 11.9 Hz, 2H), 1.11 (d, J = 21.8 Hz, 12H). IR (KBr disk, cm−1): 3075 (w), 2975 (m), 2940 (w), 1745 (s), 1641 (s), 1595 (m), 1468 (s), 1374 (m), 1214 (s), 964 (w), 795 (m), 720 (w).Synthesis of 1-(2-((1-oxyl-2,2,6,6-tetramethylpiperidin-4-yl)oxy)-2-oxoethyl)-4-(3-(pyridin-2-yl)-1H-pyrazol-1-yl)pyridin-1-ium (1d)
Creamcolored solid of 1d was obtained by the similar method to that used for the isolation of 1a, using 2,2,6,6-tetramethylpiperidinooxy-4-yl 2-iodoacetate (0.55 g, 1.6 mmol) and 2-(1-(pyridin-4-yl)-1H-pyrazol-3-yl)pyridine (0.30 g, 1.4 mmol) as starting materials. Yield: 0.53 g (70%). HRMS calcd for C24H29N5O3+: 435.2265; found: 435.2282. Anal. calcd for C24H29N5O3I: C, 51.25; H, 5.20; N, 12.45%. Found: C, 50.99; H, 5.32; N, 12.08%. 1H NMR (400 MHz, CDCl3): δ 9.10 (d, J = 6.0 Hz, 2H), 8.54 (d, J = 3.8 Hz, 1H), 8.41 (s, 1H), 8.27 (d, J = 6.3 Hz, 2H), 7.95 (d, J = 7.8 Hz, 1H), 7.67 (t, J = 7.2 Hz, 1H), 7.19 (d, J = 2.3 Hz, 2H), 5.93 (s, 2H), 5.14 (d, J = 10.5 Hz, 1H), 2.07 (d, J = 11.7 Hz, 2H), 1.87 (t, J = 11.2 Hz, 2H), 1.22 (d, J = 41.2 Hz, 12H). IR (KBr disk, cm−1): 2972 (m), 2950 (w), 1753 (s), 1641 (s), 1525 (s), 1483 (m), 1373 (s), 1288 (w), 1211 (s), 952 (m), 775 (w).Typical procedure for the formation of aldehydes from alcohols
A test tube equipped with a magnetic stirring bar was charged with CuBr (14.4 mg, 0.10 mmol, 5 mol%), 1a (70.6 mg, 0.12 mmol) and Na2CO3 (10.6 mg, 0.10 mmol). The reaction vessel was vacuumed and backfilled with oxygen for three times. Then 4-methylbenzyl alcohol (0.24 g, 2.0 mmol) and [bmim][BF4] (3 mL) were added. The resulting solution was stirred under an oxygen balloon at 50 °C (or 70 °C for (4-nitrophenyl)methanol, (4-trifluorophenyl)methanol, (4-bromo-phenyl)methanol and (4-chlorophenyl)methanol) for 24 h. Then it was extracted three times with Et2O (3 × 5 mL). The combined organic layer was washed with brine (20 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography using petroleum ether and ethyl acetate as the eluent.Typical procedure for the formation of imines from alcohols
A test tube equipped with a magnetic stirring bar was charged with CuBr (14.4 mg, 0.10 mmol), 1a (70.6 mg, 0.12 mmol) and Na2CO3 (10.6 mg, 0.10 mmol). The reaction vessel was vacuumed and backfilled with oxygen for three times. And then 4-methylbenzyl alcohol (0.24 g, 2.0 mmol), aniline (0.22 g, 2.4 mmol) and [bmim][BF4] (3 mL) were added subsequently. The resulting solution was stirred under an oxygen balloon at 80 °C (or 100 °C for (4-nitrophenyl)methanol, (4-trifluorophenyl)methanol and (4-chlorophenyl)methanol) for 24 h. Then it was cooled to ambient temperature, and extracted three times with Et2O (3 × 5 mL). The combined organic layer was washed with brine (20 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash column chromatography using petroleum ether and ethyl acetate as the eluent.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21373142 and 21471108 and 21531006), the State Key Laboratory of Organometallic Chemistry of Shanghai Institute of Organic Chemistry (2015kf-07), the “333” Project of Jiangsu Province, the Priority Academic Program Development of Jiangsu Higher Education Institutions, the “SooChow Scholar” Program of Soochow University.Notes and references
- S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400–3420 CrossRef PubMed.
- R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245–251 CrossRef.
- E. Lindbäck, S. Dawaigher and K. Wärnmark, Chem.–Eur. J., 2014, 20, 13432–13481 CrossRef PubMed.
- (a) R. A. Sheldon, Catal. Today, 2015, 247, 4–13 CrossRef; (b) R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) Oxidation of Alcohols to Aldehydes and Ketones, A Guide To Current Practice, ed. G. Tojo and M. I. Fernandez, Springer, Berlin, 2006 Search PubMed.
- (a) T. Nishimura, T. Onoue, K. Ohe and S. Uemura, J. Org. Chem., 1999, 64, 6750–6755 CrossRef CAS PubMed; (b) M. J. Schultz, S. S. Hamilton, D. R. Jensen and M. S. Sigman, J. Org. Chem., 2005, 70, 3343–3352 CrossRef CAS PubMed; (c) M. S. Sigman and D. R. Jensen, Acc. Chem. Res., 2006, 39, 221–229 CrossRef CAS PubMed; (d) G. Urgoitia, A. Maiztegi, R. SanMartin, M. T. Herrero and E. Domínguez, RSC Adv., 2015, 5, 103210–103217 RSC.
- (a) S. Chibani, C. Michel, F. Delbecq, C. Pinel and M. Besson, Catal. Sci. Technol., 2013, 3, 339–350 RSC; (b) T. Mallat and A. Baiker, Appl. Catal., A, 2000, 200, 3–22 CrossRef CAS; (c) I. Reile, A. Paju, M. Eek, T. Pehk and M. Lopp, Synlett, 2008, 347–350 CAS.
- (a) A. Dijksman, A. Marino-González, A. M. i. Payeras, I. W. C. E. Arends and R. A. Sheldon, J. Am. Chem. Soc., 2001, 123, 6826–6833 CrossRef CAS PubMed; (b) R. A. Sheldon and I. W. C. E. Arends, J. Mol. Catal. A: Chem., 2006, 251, 200–214 CrossRef CAS; (c) H. Mizoguchi, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2014, 53, 3178–3182 CrossRef CAS PubMed.
- (a) F. Saleem, G. K. Rao, A. Kumar, G. Mukherjee and A. K. Singh, Organometallics, 2014, 33, 2341–2351 CrossRef CAS; (b) O. Prakash, K. N. Sharma, H. Joshi, P. L. Gupta and A. K. Singh, Organometallics, 2014, 33, 983–993 CrossRef CAS.
- (a) M. L. Personick, B. Zugic, M. M. Biener, J. Biener, R. J. Madix and C. M. Friend, ACS Catal., 2015, 5, 4237–4241 CrossRef CAS; (b) S. Wang, J. Wang, Q. F. Zhao, D. D. Li, J. Q. Wang, M. Cho, H. Cho, O. Terasaki, S. J. Chen and Y. Wan, ACS Catal., 2015, 5, 797–802 CrossRef CAS; (c) W. Y. Zhou, P. Tian, F. A. Sun, M. Y. He and Q. Chen, J. Catal., 2016, 335, 105–116 CrossRef CAS; (d) A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403–1412 CrossRef CAS PubMed.
- (a) L. Y. Wang, S. S. Shang, G. S. Li, L. H. Ren, Y. Lv and S. Gao, J. Org. Chem., 2016, 81, 2189–2193 CrossRef CAS PubMed; (b) H. Bataineh, O. Pestovsky and A. Bakac, ACS Catal., 2015, 5, 1629–1637 CrossRef CAS; (c) J. X. Liu and S. M. Ma, Org. Lett., 2013, 15, 5150–5153 CrossRef CAS PubMed; (d) S. S. Kotha, S. Chandrasekar, S. Sahu and G. Sekar, Eur. J. Org. Chem., 2014, 7451–7457 CrossRef CAS.
- (a) J. L. Long, S. B. Wang, Z. X. Ding, S. C. Wang, Y. G. Zhou, L. Huang and X. X. Wang, Chem. Commun., 2012, 48, 11656–11658 RSC; (b) B. L. Ryland and S. S. Stahl, Angew. Chem., Int. Ed., 2014, 53, 8824–8838 CrossRef CAS PubMed.
- M. F. Semmelhack, C. R. Schmid, D. A. Cortes and C. S. Chou, J. Am. Chem. Soc., 1984, 106, 3374–3376 CrossRef CAS.
- (a) M. M. Hossain and S. G. Shyu, Adv. Synth. Catal., 2010, 352, 3061–3068 CrossRef CAS; (b) L. Tebben and A. Studer, Angew. Chem., Int. Ed., 2011, 50, 5034–5068 CrossRef CAS PubMed; (c) Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 RSC; (d) S. Wertz and A. Studer, Green Chem., 2013, 15, 3116–3134 RSC; (e) N. Jiang and A. J. Ragauskas, J. Org. Chem., 2006, 71, 7087–7090 CrossRef CAS PubMed.
- (a) J. Y. Hou, Y. Luan, J. Tang, A. M. Wensley, M. Yang and Y. F. Lu, J. Mol. Catal. A: Chem., 2015, 407, 53–59 CrossRef CAS; (b) A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ACS Catal., 2011, 1, 48–53 CrossRef CAS.
- (a) D. W. Tan, J. B. Xie, Q. Li, H. X. Li, J. C. Li, H. Y. Li and J. P. Lang, Dalton Trans., 2014, 43, 14061–14071 RSC; (b) J. B. Xie, J. J. Bao, H. X. Li, D. W. Tan, H. Y. Li and J. P. Lang, RSC Adv., 2014, 4, 54007–54017 RSC.
- (a) B. Karimi, A. Biglari, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2007, 46, 7210–7213 CrossRef CAS PubMed; (b) T. Fey, H. Fischer, S. Bachmann, K. Albert and C. Bolm, J. Org. Chem., 2001, 66, 8154–8159 CrossRef CAS PubMed; (c) C. Aellig, D. Scholz, S. Conrad and I. Hermans, Green Chem., 2013, 15, 1975–1980 RSC; (d) H. Zhang and L. L. Fu, Synth. Commun., 2014, 44, 610–619 CrossRef CAS; (e) L. Di and Z. Hua, Adv. Synth. Catal., 2011, 353, 1253–1259 CrossRef CAS.
- (a) A. Gheorghe, T. Chinnusamy, E. Cuevas-Yañez, P. Hilgers and O. Reiser, Org. Lett., 2008, 10, 4171–4174 CrossRef CAS PubMed; (b) A. P. Dobbs, P. Jones, M. J. Penny and S. E. Rigby, Tetrahedron, 2009, 65, 5271–5277 CrossRef CAS; (c) A. Gheorghe, E. Cuevas-Yanez, J. Horn, W. Bannwarth, B. Narsaiah and O. Reiser, Synlett, 2006, 2767–2770 CAS.
- (a) Y. Huangfu, Q. Sun, S. X. Pan, X. J. Meng and F. S. Xiao, ACS Catal., 2015, 5, 1556–1559 CrossRef; (b) J.-P. Lindner, C. Röben, A. Studer, M. Stasiak, R. Ronge, A. Greiner and H.-J. Wendorff, Angew. Chem., Int. Ed., 2009, 48, 8874–8877 CrossRef CAS PubMed; (c) T. Y. S. But, Y. Tashino, H. Togo and P. H. Toy, Org. Biomol. Chem., 2005, 3, 970–971 RSC.
- (a) A. Schätz, R. N. Grass, W. J. Stark and O. Reiser, Chem.–Eur. J., 2008, 14, 8262–8266 CrossRef PubMed; (b) A. K. Tucker-Schwartz and R. L. Garrell, Chem.–Eur. J., 2010, 16, 12718–12726 CrossRef CAS PubMed; (c) B. Karimi and E. Farhangi, Chem.–Eur. J., 2011, 17, 6056–6060 CrossRef CAS PubMed.
- (a) W. X. Qian, E. L. Jin, W. L. Bao and Y. M. Zhang, Tetrahedron, 2006, 62, 556–562 CrossRef CAS; (b) C. J. Zhu, L. Ji and Y. Y. Wei, Catal. Commun., 2010, 11, 1017–1020 CrossRef CAS; (c) H. A. Beejapur, F. Giacalone, R. Noto, P. Franchi, M. Lucarini and M. Gruttadauria, ChemCatChem, 2013, 5, 2991–2999 CrossRef CAS; (d) R. J. Hu, M. Lei, H. G. Wei and Y. G. Wang, Chin. J. Chem., 2009, 27, 587–592 CrossRef CAS; (e) C. X. Miao, L. N. He, J. L. Wang and F. Wu, J. Org. Chem., 2010, 75, 257–260 CrossRef CAS PubMed; (f) X. L. Liu, Q. Q. Xia, Y. J. Zhang, C. Y. Chen and W. Z. Chen, J. Org. Chem., 2013, 78, 8531–8536 CrossRef CAS PubMed; (g) J. K. Kim, A. Matic, J. H. Ahn, P. Jacobsson and C. E. Song, RSC Adv., 2012, 2, 10394–10399 RSC; (h) Y. C. Zhang, F. L. Lü, X. H. Cao and J. Q. Zhao, RSC Adv., 2014, 4, 40161–40169 RSC; (i) X. E. Wu, L. Ma, M. X. Ding and L. X. Gao, Synlett, 2005, 607–610 CAS; (j) A. Falla, M. Senea, M. Gayeb, G. Gómeza and Y. Fall, Tetrahedron Lett., 2010, 51, 4501–4504 CrossRef; (k) C. J. Zhu, A. Yoshimura, Y. Y. Wei, V. N. Nemykin and V. V. Zhdankin, Tetrahedron Lett., 2012, 53, 1438–1444 CrossRef CAS; (l) L. Liu, J. J. Ma, L. Y. Ji and Y. Y. Wei, J. Mol. Catal. A: Chem., 2008, 291, 1–4 CrossRef CAS.
- Z. G. Wang, Y. Jin, X. H. Cao and M. Lu, New J. Chem., 2014, 38, 4149–4154 RSC.
- C. W. Y. Chung and P. H. Toy, J. Comb. Chem., 2007, 9, 115–120 CrossRef CAS PubMed.
- B. T. Chen, K. V. Bukhryakov, R. Sougrat and V. O. Rodionov, ACS Catal., 2015, 5, 1313–1317 CrossRef CAS.
- L. C. Li, R. Matsuda, I. Tanaka, H. Sato, P. Kanoo, H. J. Jeon, M. L. Foo, A. Wakamiya, Y. Murata and S. Kitagawa, J. Am. Chem. Soc., 2014, 136, 7543–7546 CrossRef CAS PubMed.
- I. A. Ansari and R. Gree, Org. Lett., 2002, 4, 1507–1509 CrossRef CAS PubMed.
- N. Jiang and A. J. Ragauskas, Org. Lett., 2005, 7, 3689–3692 CrossRef CAS PubMed.
- F. L. Zeng and Z. K. Yu, J. Org. Chem., 2006, 71, 5274–5281 CrossRef CAS PubMed.
- J. C. Li, H. X. Li, H. Y. Li, W. J. Gong and J. P. Lang, Cryst. Growth Des., 2016, 16, 1617–1625 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The 1H and 13CNMR data of aldehyde and imine products, the 1H and 13C NMR spectra of 4-(2-iodineacetoxy)-2,2,6,6-tetramethylpiperidine 1-oxyl, 1a–1d, 2b, aldehyde and imine products, and the positive-ion ESI mass spectra of 1a–1d. See DOI: 10.1039/c6ra10373a |
| This journal is © The Royal Society of Chemistry 2016 |