
Co2Mo3O8/reduced graphene oxide composite: synthesis, characterization, and its role as a prospective anode material in lithium ion batteries†
Sandeep K. Marka‡
ab,
Shaikshavali Petnikota‡ab,
Vadali V. S. S. Srikanth*a,
M. V. Reddy*bc,
Stefan Adamsc and
B. V. R. Chowdarib
aSchool of Engineering Sciences and Technology (SEST), University of Hyderabad, Gachibowli, Hyderabad 500046, India. E-mail: vvsssse@uohyd.ernet.in; Tel: +91 40 2313 4453
bDepartment of Physics, National University of Singapore, Singapore 117542, Singapore. E-mail: phymvvr@nus.edu.sg; msemvvr@nus.edu.sg; reddymvvr@gmail.com; Tel: +65 6516 2605
cDepartment of Materials Science and Engineering, National University of Singapore, Singapore 117576, Singapore
First published on 1st June 2016
Abstract
A Co2Mo3O8/reduced graphene oxide (Co2Mo3O8/rGO) composite was synthesized by following a single step solid state reduction procedure. The prepared Co2Mo3O8/rGO composite was characterized using a multitude of characterization techniques, which confirmed the formation of the composite. Electron micrographs clearly showed that the composite consisted of submicron sized (lateral) and 50 nm thick hierarchical hexagonal nanoplatelets of Co2Mo3O8 attached to thin graphene layers of rGO. Raman scattering analysis not only confirmed the presence of Co2Mo3O8 and rGO in the composite but also revealed that the defects present in rGO are more than that in GO. Through thermogravimetric analysis, the amount of rGO present in the composite was found to be ∼22% by weight. Co 2p, Mo 3d, C 1s and O 1s X-ray photoelectron energy peaks were clearly identified. The analysis of these peaks confirmed the oxidation states of the respective elements in stoichiometric Co2Mo3O8. The as-synthesized Co2Mo3O8/rGO composite was tested as an anode material in half-cell configured lithium ion batteries. When cycled at 60 mA g−1 current density and in the 0.005–3.0 V range, the Co2Mo3O8/rGO composite delivered an excellent reversible specific capacity of ∼954 mA h g−1 that corresponds to 82% capacity retention at the end of the 60th cycle, which is higher than the theoretical capacity of both Co2Mo3O8 and graphene. Moreover, the Co2Mo3O8/rGO composite exhibited excellent rate capability. A reversible specific capacity of 471 mA h g−1 (at a current density of 1000 mA g−1) was delivered at the end of the 31st cycle. The value increased to 1006 mA h g−1 when the current density was switched to 100 mA g−1 at the end of the 36th cycle. Redox peaks in the cyclic voltammetry (CV) curves revealed that electrochemical conversion and electrochemical adsorption and desorption type reaction mechanism are the primary reasons for lithium ion storage. A constant area under the CV curves throughout the tests was noticed, which is an indication of the stable capacity while the CV results are in line with the galvanostatic cycling (GC) results. From the CV and GC results, it is concluded that the higher specific capacity, longer cycle life, and better rate capability are due to the excellent synergy between Co2Mo3O8 and rGO in the composite.
1. Introduction
The advent of rechargeable Li-ion batteries (LIBs) made it possible to develop electric vehicles (EVs) and hybrid electric vehicles (HEVs) which help in drastically reducing pollution from vehicles. However, high energy and power density delivering rechargeable LIBs with qualities such as long cycle life, trustworthy safety, and affordable costs are required to power EVs or HEVs.1 To fulfill the above criteria, molybdenum (Mo) based oxides (MoO2 and MoO3),2,3 chalcogenides (MoS2 and MoSe2),4,5 and oxysalts (AxMoyOz, where A = Fe, Co, Ni, Ca, Mn, Zn, Mg, or Cd and x = 1, y = 1, z = 4; x = 2, y = 3, z = 8)6–14 have been developed as anode materials in LIBs. Nanosized Mo oxides showed better electrochemical properties than their bulk counterparts due to certain merits such as shortened diffusion path length for Li-ions, excellent stability against physical strains during the Li-ion cycling, and the presence of numerous active surface sites which facilitate enough electrolyte's adsorption.15 However, agglomeration of nanoparticles upon cycling has been a major problem, which leads to pulverization and cracking of the electrode and ultimately it causes capacity fading. To overcome any disadvantages, composites with carbonaceous materials such as amorphous carbon,16 one and two-dimensional17,18 carbon nanomaterials were prepared. These composites exhibited enhanced electrical conductivity and due to the synergy between the constituents of the composites, volume changes during cycling could be suppressed.19–23 Mixed metal oxides are promising anode materials for LIBs, but disadvantages such as poor electrical conductivity, slow diffusion kinetics, etc.,24–26 are associated with these materials. Of late, it was shown that Mo-cluster oxysalt/graphene composites exhibit high specific capacity values, long cycle lives, and enhanced rate capabilities.With regards to the synthesis of Mo-cluster oxysalt/graphene composites, a two-step reduction method was used to synthesize hierarchically nanostructured Mn2Mo3O8–graphene27 and Fe2Mo3O8–graphene28 nanocomposites which exhibited excellent reversible specific capacities of ∼951 and ∼835 mA h g−1, respectively, after 40 cycles in 0.01–3.0 V range and at a current density of 200 mA g−1. However, the two-step method mentioned above,27,28 involved the reaction of phosphomolybdic acid, the respective metal acetate and graphene oxide (GO) with hydrazine hydrate to form an intermediate product which was heat-treated at 550 °C for 5 h in a reducing H2/Ar atmosphere to obtain the final product. Recently we have developed a single-step reduction method to synthesize FLG–Co2Mo3O8 composite29 in which exhibited excellent reversible specific capacity of ∼785 mA h g−1 after 50 cycles in 0.005–3.0 V range and at a current density of 60 mA g−1. The single-step method mentioned,29 involved heating a mechanically mixed blend of GO, MoO3 and Co(OOCCH3)2·4H2O at 750 °C for 8 h in Ar atmosphere. Even though FLG–Co2Mo3O8 composite exhibited excellent reversible specific capacity, some drawbacks such as continuous capacity fading at higher current densities, high irreversible capacity loss (ICL) and low coulombic efficiency were associated with the composite.29 The main reason for the drawbacks was identified as the lack of optimal synergy between FLG and Co2Mo3O8 platelets, and improper processing conditions were identified as the reason for the same. Given this, the reaction chemistry was altered, and excellent synergy between graphene layers and Co2Mo3O8 platelets was established. As a result, the Co2Mo3O8/rGO composite, when tested as an anode material in LIBs, exhibited excellent electrochemical properties, enhanced specific capacity, long cycle life, and better rate capability unlike the FLG–Co2Mo3O8 composite29 reported in our previous work.
2. Experimental work
2.1 Synthesis of materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:2 molar ratio and mixed using mechanical grinding for 30 min. The molecular formula of GO was taken as C2.2H0.8O1 (ref. 30) for molar calculations. Then the reaction mixture (12 g) was placed into a ceramic boat and transferred to a tubular furnace (Carbolite, UK). Thermal annealing of the reaction mixture was carried out at 750 °C for 8 h. The furnace was then cooled to room temperature at a cooling rate of 5 °C min−1, and a black colored fine powder (6 g) was collected. Heating was started after flushing out air in the furnace with Ar gas while Ar gas flow was maintained throughout the experiment. The as-synthesized final product was directly (i.e., without any further purification or functionalization/modification) used for characterization and electrochemical analysis.
:3:2 molar ratio and mixed using mechanical grinding for 30 min. The molecular formula of GO was taken as C2.2H0.8O1 (ref. 30) for molar calculations. Then the reaction mixture (12 g) was placed into a ceramic boat and transferred to a tubular furnace (Carbolite, UK). Thermal annealing of the reaction mixture was carried out at 750 °C for 8 h. The furnace was then cooled to room temperature at a cooling rate of 5 °C min−1, and a black colored fine powder (6 g) was collected. Heating was started after flushing out air in the furnace with Ar gas while Ar gas flow was maintained throughout the experiment. The as-synthesized final product was directly (i.e., without any further purification or functionalization/modification) used for characterization and electrochemical analysis.2.2 Characterization of materials
Morphological studies were carried out by using field emission scanning electron microscope (FESEM, model Ultra 55 Carl Zeiss) and transmission electron microscope (TEM, model FEI Technai G2 S-Twin) operated at 5 and 200 kV, respectively. The crystallinity of the samples was studied using the X-ray diffraction (XRD) technique with Bruker AXS model D8 Advanced system. XRD patterns were recorded from 10 to 100° using Cu Kα as the X-ray source (λ = 1.54 Å). Raman spectra were collected in the air and at room temperature with (Renishaw Raman system 2000) auto-excitation wavelength with 1 μm spot size focused by 50× objective lens. Specific surface area and porosity distribution of the samples were measured by performing N2 physisorption (on Micromeritics, model Tristar 3000) at 77 K using Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) multipoint methods. Samples were preheated under N2 flow for 1 h at 180 °C. While recording N2 adsorption and desorption isotherms, NTP conditions were maintained around the sample holder, and a mixture of He and N2 gasses was flown-in according to the dynamic (flowing gas) technique.To know the amount of graphene in the composite, oxidative decomposition of the Co2Mo3O8/rGO composite was performed in the temperature range 25–1000 °C at 10 °C min−1 heating rate by using TA Instrument 2960 (DTA-TGA). X-ray photoelectron spectroscopy (XPS, AXIS Ultra DLD spectrometer (Kratos Analytica) with monochromatic Al Kα radiation) measurements were carried out on the synthesized composite to estimate different bonds and their binding energy values present in the composite. XPS data was analyzed by using Casa XPS software. Survey spectra were recorded in the energy range 0–1200 eV. Charge referencing was carried out against carbon C (C 1s binding energy = 284.6 eV).
2.3 Battery fabrication and testing
Electrode slurry was prepared by uniformly mixing Co2Mo3O8/rGO composite material, polyvinylidene fluoride (PVDF, Kynar 2801) polymer binder and Super-P carbon black (ENSACO, MMM Super P, 230 m2 g−1) (in 70:15:15 ratio by weight) in N-methyl pyrrolidinone solvent at ambient conditions on a magnetic stirrer for 12 h. So-prepared electrode slurry was then coated as ∼25 μm thick film onto an etched Cu foil (thickness ∼10 μm) by using ‘doctor blade’ technique. After 12 h drying at 80 °C in a hot air oven, the electrode foil was pressed using stainless steel roller. Subsequently, the foil was cut into 16 mm diameter circular discs. The geometrical electrode area was around 2 cm2, and mass of active material was estimated to be 2–4 mg (70% of overall electrode mass excluding Cu foil weight). All constituents of the battery namely bottom and top caps, electrodes, and separator were dried in vacuum oven at 80 °C for 12 h before pressing the coin cell type batteries. Coin cells CR2016 were assembled in Ar gas filled glove box (MBraun) by using the fabricated electrodes as anodes and Li metal (16 mm diameter and 0.59 mm thickness, Hohsen Corp.) as the counter electrode. Celgard 2502 polymer membrane was used as the separator while 1 M LiPF6 in ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:1 by volume, Merck Selectipur LP40) was used as the electrolyte. The fabricated batteries were tested at room temperature with cyclic voltammetry (CV, MacPile II, Biologic) and galvanostatic cycling (GC, model SCN, Bitrode). The cells were examined in the voltage range 0.005–3.0 V at a scan rate of 58 μV s−1 in CV study while GC test was carried out in the same voltage window as in CV and at different current rates namely 60, 100, 200, 400, 600, 800 and 1000 mA g−1. Here it should be noted that a slow scan rate of 0.058 mV s−1 is used to test cathode31–35 and anode materials36–42 because at this scan rate materials present even in minute quantities participate in electrochemical cycling and in general, CV peaks are well resolved. On the other hand, high scan rates have resulted in broadening of peaks, shifting of anodic peaks to higher voltages and cathodic peaks to lower voltages, and increasing the peak current values.43–46 Electrochemical impedance spectroscopic (EIS) measurements of the coin cells were done with Solartron impedance/gain-phase analyzer (model SI 1255) coupled with a potentiostat (SI 1268) at room temperature in the frequency range 180000 to 0.003 Hz with an AC signal amplitude of 10 mV. The acquired impedance data was analyzed using Z-view software (version 2.2, Scribner Assoc., Inc.).
3. Results and discussion
3.1 Morphological, structural, phase and adsorption analyses
FESEM images of the as-synthesized composite are shown Fig. 1. The images clearly show that the composite consisted of submicron sized (lateral) and ∼50 nm thick hierarchical hexagonal nanoplatelets of Co2Mo3O8 attached to thin graphene layers of rGO similar to the observations in our previous work.29 However, the high magnification images (Fig. 1(b)–(d)) show the nanoplatelets are more uniformly distributed and are entrenched between the graphene layers unlike in our previous work.29 Such an architecture is expected to be beneficial when the material is tested as an anode in LIBs because the conducting graphene sheets facilitate much easier adsorption of electrolyte which leads to better conduction of Li-ions and electrons in the composite upon cycling. The contribution of Super P carbon to the electrochemical specific capacity is negligible but it provides good contact between neighboring regions in Co2Mo3O8/rGO composite (please see ESI, Fig. ESI1†). | ||
| Fig. 1 FESEM images of Co2Mo3O8/rGO composite at different magnifications. | ||
The morphology was further confirmed by analyzing TEM images which are shown in Fig. 2. Here it is very important to note that even after rigorous sonication during TEM sample preparation, Co2Mo3O8 particles do not lose their contact with the graphene layers. TEM images clearly show that graphene sheets and some of the Co2Mo3O8 nanoplatelets are semi-transparent to the electron beam. The thickness of the Co2Mo3O8 nanoplatelets as measured from the TEM images was ∼55 nm, a value close to that measured from FESEM images. TEM images also show that each of the Co2Mo3O8 nanoplatelets is attached to the graphene sheets which are observed to be wrinkled, a case typical to that of rGO.
 | ||
| Fig. 2 TEM images of Co2Mo3O8/rGO composite at different magnifications. | ||
The diffraction peaks in the XRD pattern (Fig. 3(a)) of the composite could be indexed (also matched with the peaks corresponding to the JCPDS data file no. 34-0511 for Co2Mo3O8 phase) to the hexagonal crystal structure of Co2Mo3O8. Rietveld refined XRD data confirmed the formation of the composite. The refined data showed that 81.3% of it corresponded to the phase pure Co2Mo3O8 while the remaining 18.7% corresponded to the combination of intense (002) and broad (004) diffraction peaks of rGO, and monoclinic MoO2 phase (JCPDS file number #76-1807). (011) and (022) diffraction peaks of monoclinic MoO2 appeared at 26.5° and 53.6°, respectively. Rietveld refinement showed that Co2Mo3O8 nanoplatelets have crystallized in hexagonal phase with lattice parameters a = 5.785(8) Å and c = 9.923(5) Å. The strong characteristic diffraction peak at 26.5° and a broad diffraction peak at 53.6° (both indicated with an asterisk in the Fig. 3(a)) correspond to (002) and (004) graphite planes in rGO.
 | ||
| Fig. 3 (a) X-ray diffractogram and (b) Raman spectrum of Co2Mo3O8/rGO composite, respectively. | ||
Raman scattering analysis further confirmed the presence of rGO in the composite. The appearance of characteristic Raman bands (as shown in Fig. 3(b)) namely the D band at ∼1345 cm−1, the G band at ∼1588 cm−1 and the 2D band centered at ∼2700 cm−1 (please see Fig. ESI2†) confirmed the presence of rGO47 in the composite. The intensity ratio ID/IG in the case of Co2Mo3O8/rGO is ∼0.92 which is greater than ∼0.68 measured in the case of GO, suggesting the presence of more defects in the case of rGO in the composite than in the case of GO. On the other hand, the higher intensity of G band in comparison to that of D band in both rGO and GO cases indicates the presence of good-quality regular carbon hexagons in the material.47 Raman bands related to Co2Mo3O8 in the composite are shown in Fig. ESI2.†
Co2Mo3O8/rGO composite's N2 adsorption and desorption isotherm is shown in Fig. 4(a). It is representative of the type IV isotherm of IUPAC standard adsorption–desorption isotherms. This observation is similar to that of our previous report.29 BET specific surface area and average pore diameter values of the composite powder are 63.8 m2 g−1 and 11.1 nm, respectively, which are higher than those reported previously for a similar composite.29 Fig. 4(b) shows the thermal decomposition characteristics of the composite. It shows that there is a slight weight loss below 350 °C. This loss is attributed to the evaporation of physisorbed water molecules and the decomposition of some residual oxygen-containing groups on the rGO. A rapid weight loss at ∼462 °C is attributed to combustion of carbon skeleton in the rGO.48 Unlike in our previous report,29 the oxidation of Co2Mo3O8 is not observed here in the entire temperature range. This is an indication that Co2Mo3O8 nanoplatelets are covered by graphene layers as indicated in by the electron micrographs. From the weight loss characteristics, the calculated weight fraction of rGO in the Co2Mo3O8/rGO composite is ∼22 wt%.
 | ||
| Fig. 4 (a) N2 adsorption and desorption isotherm and (b) weight loss versus temperature (TGA) and derivative weight loss versus temperature (DTA) curves of Co2Mo3O8/rGO composite, respectively. | ||
XPS measurements are carried out to evaluate the precise bonding nature in Co2Mo3O8/rGO composite. The high-resolution Co 2p photoelectron spectrum shown in Fig. 5(a) displays the characteristic Co2+ peaks at 778.4 eV and 794.0 eV which are ascribed to 2p3/2 and 2p1/2 levels, respectively. Two characteristic satellite peaks of Co2+ are identified at 783.8 and 800.1 eV, and the appearance of these peaks is due to the charge transfer from ligand to the metal during the photoemission process.49 Similarly, as shown in Fig. 5(b) the spectrum of Mo 3d contains characteristic doublet at 230.5 (3d5/2) and 233.6 (3d3/2) eV corresponding to Mo4+ oxidation state. In addition to Mo4+ peaks, Mo6+ peaks are also found at 232.3 and 235.5 eV.50 As shown in Fig. 5(c), the characteristic C 1s spectrum of the composite is deconvoluted into two peaks. The peak at 284.5 eV is assigned to either sp2 or sp3 hybridized carbon atoms in the graphene, and the peak at 285.5 eV arises from C–OH bond in the graphene's basal plane. The spectrum of O 1s shown in Fig. 5(d) contain two peaks at 530.9 and 532.2 eV which correspond to oxygen atom bonded to the lattice and the surface impurities, respectively.51 All in all, the XPS analysis confirms the oxidation states of Co and Mo as 2+ and 4+, respectively. The observed Mo6+ state is speculated to be either due to surface oxidation of Co2Mo3O8 or change in oxygen co-ordination due to laser degradation process. However, any features of Mo6+ (MoO3) state are neither identified in XRD analysis nor in CV (redox couple at 2.5 V) analysis.
 | ||
| Fig. 5 X-ray photoelectron spectra of Co2Mo3O8/rGO composite: (a) Co 2p peak, (b) Mo 3d peak, (c) C 1s peak and (d) O 1s peak. | ||
Owing to the above-discussed characteristics, it is expected that there is an easy electrolyte access to the electrode material, and there are low contact and charge transfer impedances and short transport lengths for both Li-ions and electrons when Co2Mo3O8/rGO composite is tested as an anode material in LIBs.
3.2 Electrochemical analysis
A series of electrochemical measurements were carried out to test the potential usage of the Co2Mo3O8/rGO composite as anode material LIBs. Fig. 6(a) shows the CV characteristics of Co2Mo3O8/rGO composite during the first six cycles of both cathodic and anodic scans in the voltage range of 0.005–3.0 V and at a scan rate of 58 μV s−1. The CV curves are subtly different than those reported previously.29 In the first cathodic scan, two primary reduction peaks are observed, a peak at 0.6 V and a sudden rapid increase in the cathodic current leading to the appearance of a broad peak in between 0.5 and 0.005 V. Similarly in the first anodic scan, two oxidation peaks at ∼1.43 and ∼1.78 V are assigned to oxidation of Co0 and Mo0 metal nanoparticles (formed during the 1st cathodic scan) to metal oxides, respectively. Asymmetry in the CV curves indicates irreversibility in the Li ions' cycling which resulted in less coulombic efficiency (∼64%, as shown in Fig. 6(c)) in the first cycle. From the 2nd cathodic scan onwards, reduction peaks in the 1st cathodic scan have shifted towards higher voltages (i.e., 0.7 and 0.16 V, respectively and also attained symmetry) with constant reduction currents. New reduction peaks which correspond to Li ions uptake into both rGO and Co2Mo3O8 lattice have also appeared.14 The large current difference between 1st and 2nd cathodic scans and shift in peak positions are expected due to the combined effect of irreversible lithium insertion in the lattice, electrolyte decomposition, solid electrolyte interphase,29 and conversion of Co2+ and Mo4+ to their metallic states and the formation of amorphous Li2O phase as shown in eqn (1).| Co2Mo3O8 + 16Li+ + 16e− → 2Co0 + 3Mo0 + 8Li2O | (1) |
 | ||
| Fig. 6 (a) Cyclic voltammetry curves at different cycles in the voltage range 0.005–3.0 V vs. Li and at a scan rate of 58 μV s−1, (b) galvanostatic discharge and charge curves at 1st, 2nd and 60th cycles in the voltage range 0.005–3.0 V vs. Li and at a current density of 60 mA g−1, (c) discharge–charge specific capacity and coulombic efficiency vs. cycle number and (d) rate capability in the voltage range 0.005–3.0 V vs. Li of Co2Mo3O8/rGO composite. | ||
In this study, from the 2nd cycle onwards, in the cathodic scan, another two broad new reduction peaks centered at 1.30 and 1.70 V are observed, unlike our previous report.29 The appearance of these peaks is an indication of an excellent reduction of precursor materials to Co2Mo3O8/rGO composite without impurities. The redox couples at 1.70/1.80 and 1.30/1.45 V correspond to the phase transition of MoO2 due to partially lithiated LixMoO2 phase, i.e., from monoclinic phase to orthorhombic phase and vice versa during Li insertion and extraction processes, respectively.15 Evolution of the two extra peaks form the 2nd cathodic scan onwards might have originated due to the formation of MoO2 formed during the 1st cycle through Co2Mo3O8 crystal structure destruction during Li insertion (1st reduction) that eventually lead to the formation of metal nanoparticles surrounded by Li2O phase (during 1st discharge) and conversion of these metal nanoparticles to metal oxides (during 1st oxidation) as per eqn (1). Interestingly, it is noticed that compared with our previous report29 from the 2nd cycle onwards, the area under the redox curves and intensity of redox peaks are constant throughout the test. This is an indication of stable cyclability which is expected due to the presence of good cooperation between Co2Mo3O8 nanoplatelets and graphene layers in the composite material.
Further, to test the composite's cyclability and rate capability, and to support CV observations GC tests were carried out. Fig. 6(b) shows the 1st, 2nd and 60th discharge and charge cycles of Co2Mo3O8/rGO composite in the voltage range of 0.005–3.0 V and at a current density of 60 mA g−1. In the GC test, it is observed that the composite has delivered an excellent specific capacity of ∼1834 mA h g−1 during the first discharging scan from the open circuit voltage (i.e., ∼2.3 V, as shown in Fig. 6(b)) to a lower cutoff voltage of 0.005 V and as a result of this, a total of 33 Li+ ions are stored in the electrode. This specific capacity value ∼1834 mA h g−1 is much higher than the theoretical capacity14,29 value ∼873 mA h g−1 of the Co2Mo3O8/rGO composite (theoretical specific capacity of the composite = 78% (theoretical specific capacity of the Co2Mo3O8)14 + 22% (theoretical specific capacity of the rGO)52). The expected high value of specific capacity is supported by the slope change between 0.9 and 0.3 V accompanied by small voltage plateaus and after that a continuous decrease in voltage till the lower cutoff voltage 0.005 V is reached. Similarly at the end of first charging curve a reversible specific capacity of ∼1170 mA h g−1 is delivered by the electrode through gradual increase in the voltage (which is started from lower cutoff voltage 0.005 V) and a sudden change in slope at ∼1.8 V and thereafter continuous increase in the voltage till it reaches its upper cutoff voltage 3.0 V. As shown in Fig. 6(c), at the end of the first cycle ∼64% of coulombic efficiency (QE) is recorded. This whole cycle resulted in an irreversible capacity loss (ICL) of 664 mA h g−1 which is equal to a loss of ∼12 Li/Li+ ions (it is in support with observed asymmetry in the CV curves). ICL is expected due to an irreversible transfer of Li/Li+ ions by the formation of SEI film on the electrode through initial decomposition of electrolyte in the solvent.14,29,53–55 The subsequent cycles (2nd and 60th cycles) have also followed similar trend as that of the first cycle with much reduced ICL as the cycle number is increased (for example, QE of ∼98% is noticed at 30th cycle). The present study also highlights superior cycling behavior of the Co2Mo3O8/rGO composite than the other Mo3-cluster compounds reported to date. When cycled at a current density of 60 mA g−1 and in the voltage range of 0.005–3.0 V vs. Li, a reversible specific capacity of ∼954 mA h g−1 is delivered by the electrode at the end of the 60th cycle (as shown in Fig. 6(a) and (c)). The higher specific capacity and lower ICL values in this work are much better than the respective values exhibited by pure Co2Mo3O8 and Co2Mo3O8/FLG composite.14,29 The remaining discharge–charge cycles are shown in Fig. ESI3.† Further, the cycling stability of the prepared anode material is studied by conducting rate capability tests. Fig. 6(d) shows specific capacity versus cycle number profiles of Co2Mo3O8/rGO composite electrode at different current densities. The reversible specific capacities of 1001, 866, 816, 729, 628, 503 and 1006 mA h g−1 at current densities of 100, 200, 400, 600, 800, 1000 and 100 mA g−1 are measured, respectively. A minute capacity decay at higher current densities (i.e., 600 to 1000 mA g−1) might have originated from partial loss of contact between Co2Mo3O8 nanoplatelets and rGO on cycling at higher current rates. It is observed that even after returning to the current density of 100 mA g−1 from the highest current density, the discharge specific capacity was 1030 mA h g−1 (corresponding charge capacity was 1006 mA h g−1), which is an indication of the excellent stability of the electrode material even after many cycles at different current rates. The higher specific capacity, longer cycle life and better rate capability of the Co2Mo3O8/rGO composite material is expected due to the composite's architecture which facilitates good conduction in the electrode material while the composite's high surface area facilitates easy electrolyte adsorption and causes ion/electron diffusion more feasible.27,28 The excellent electrochemical properties of the present material with the available Mo3-cluster compounds are compared in Table 1.
| S. No. | Mo3-cluster compound (or composite) | Synthesis procedurea | Voltage range (V) | Reversible specific capacity (mA h g−1) @ current density (mA g−1) & cycle number |
|---|---|---|---|---|
| a CTR-carbothermal reduction, SC-solution chemistry and GTR-graphenothermal reduction. | ||||
| 1 | LiYMo3O8 (ref. 12) | CTR | 0.005–3.0 | 385 @ 30 & 120th |
| 2 | Mn2Mo3O8 (ref. 12) | CTR | 0.005–3.0 | 205 @ 30 & 50th |
| 3 | Co2Mo3O8-heat treated13 | CTR | 0.005–3.0 | 790 @ 60 & 60th |
| 4 | Cu3Mo2O9 (ref. 56) | SC | 0.01–3.0 | 754 @100 & 80th |
| 484 @ 500 & 38th | ||||
| 5 | FLG–Co2Mo3Mo8 (ref. 29) | GTR | 0.005–3.0 | 607 @ 180 & 50th |
| 6 | FLG–Mn2Mo3Mo8 (ref. 29) | GTR | 0.005–3.0 | 518 @ 180 & 50th |
| 7 | FLG–Zn2Mo3O8 (ref. 29) | GTR | 0.005–3.0 | 579 @ 180 & 50th |
| 8 | Mn2Mo3Mo8–graphene27 | SC | 0.01–3.0 | 951 @ 200 & 40th |
| 672 @ 1500 & 20th | ||||
| 9 | Fe2Mo3Mo8–graphene28 | SC | 0.01–3.0 | 835 @ 200 & 40th |
| 574.8 @ 3000 & 1st | ||||
| 10 | Present study | GTR | 0.005–3.0 | 954 @ 60 & 60th |
| 729 @ 600 & 21st | ||||
| 471 @ 1000 & 31st | ||||
EIS measurements are carried on the cycled coin cell batteries in the frequency range 180000 to 0.003 Hz to understand the Li-ion storage mechanism of the Co2Mo3O8/rGO composite and support the GC and CV observations. Fig. 7 shows Nyquist plots of the coin cell batteries at different states (i.e., at OCV, 1st discharged and 1st charged). EIS results about all the three stages of are fitted to one equivalent circuit (inset of Fig. 7), which is similar to our previous report.29
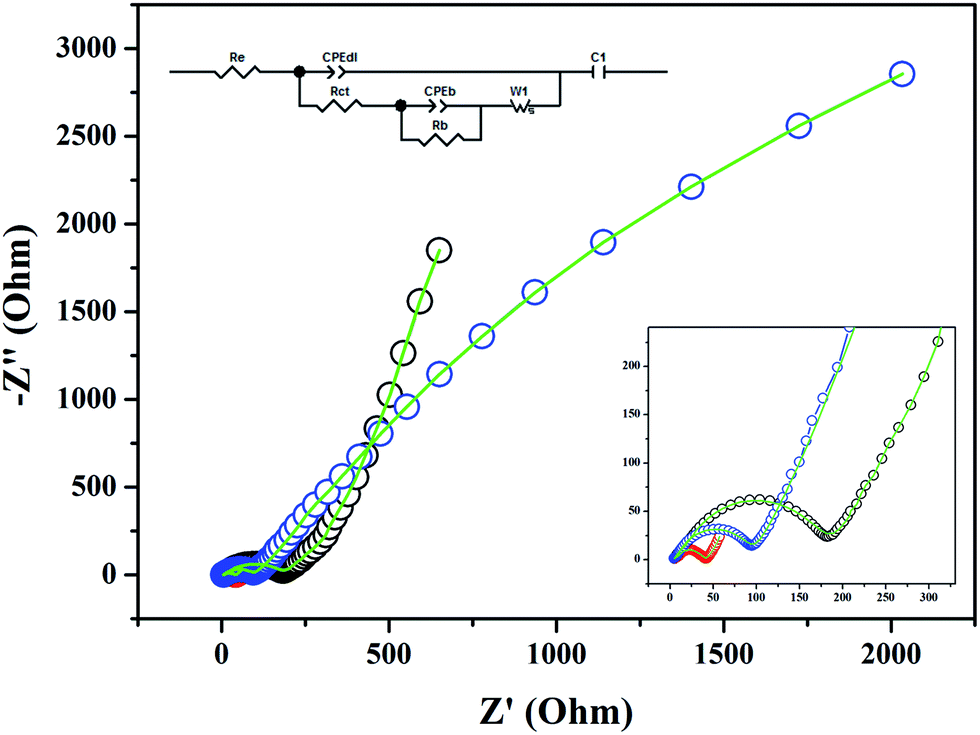 | ||
| Fig. 7 EIS curves of Co2Mo3O8/rGO composite at different charge states (black, red and blue color circles represent OCV, 1st discharge, and 1st charge scanned, respectively) with curve fitting (solid line in green color) and the inset shows the magnified portion of the EIS curves at Z′ < 300 and the equivalent electrical circuit. | ||
Certain elements (related to EIS results) that are necessary to understand the Li-ion storage kinetics and their values are tabulated in Table 2. The contact (or solvent) resistance (Re) for 1st discharge scan has decreased and slightly increased for 1st charge scan as compared to OCV. It is expected due to the formation of insulating SEI on the electrode surface during the 1st cycle. Resistance to charge transfer (Rct) in both cases is less than OCV, and it followed a decreasing trend. The lower Rct value of the 1st charge scan than the first discharge scan is an indication for easy removal of Li ions due to the presence of highly conducting and high surface area graphene, and excellent synergy between graphene and Co2Mo3O8 nanoplatelets. After the 1st discharge and charge scans, two orders of increase and one order of decrease in the electric double layer capacitance (CPEdl) values are observed, respectively, and this is expected due to formation of thick SEI film on the electrode surface while first discharge leads to form dense dielectric medium between active electrode and charge carriers in the electrolyte which causes the increase in CPEdl. A continuous decrease in the values of the bulk resistance (Rb) and bulk electric double layer capacitance (CPEb) are observed, and this is expected due to easy electrolyte access throughout the electrode to make a smooth transfer of ions or electrons in the electrode. Similar to CPEdl, intercalation capacitance (Ci) and Warburg element (Li-ion diffusion, Wi) first increased and then decreased. In all three stages, subtle values of intercalation capacity Ci57 and very high values of Warburg element Ws (resistance to Li-ion diffusion) are found when compared to Re and Rct. The very low value of Ci indicates that Li storage preferably takes place either by redox couples of Mo-clusters or electrochemical adsorption by graphene layers.56 The EIS results supported the GC and CV results in the 1st cycle and are expected to be in line with for other cycles also.
| Sample status | Re | Rct | CPEdl (μF) | Rb | CPEb (mF) | Ci (F) |
|---|---|---|---|---|---|---|
| Co2Mo3O8/rGO fresh cell | 4.6 ± 0.04 | 36.5 ± 3.8 | 39.6 ± 6.1 | 139.2 ± 3.3 | 0.02 ± 0.004 | 0.04 |
| Co2Mo3O8/rGO discharged state | 4.0 ± 0.00 | 16.5 ± 0.0 | 354.6 ± 0.0 | 85.0 ± 0.0 | 0.006 ± 0.0 | 0.05 |
| Co2Mo3O8/rGO charged state | 4.2 ± 0.00 | 4.5 ± 0.0 | 1.0 ± 0.0 | 21.4 ± 0.0 | 0.004 ± 0.0 | 0.02 |
4. Conclusions
In the present study, a single step reduction method is used to synthesize Co2Mo3O8/rGO composite. Observations on the synthesized Co2Mo3O8/rGO composite through various characterization techniques confirmed the formation of the composite. Electrochemical properties of the composite are studied with GC, CV and EIS tests. Li cycling mechanism was explained using CV in which redox couples of Co2Mo3O8 and electrochemical adsorption and desorption by rGO are found responsible for Li storage and release. In the GC test, high reversible specific capacity and longer cycle life of 954 mA h g−1 @ 60 mA g−1, at 60th cycle; excellent rate capability of 503 mA h g−1 @ 1000 mA g−1, at the 29th cycle of the electrode material are noticed. EIS test is used to probe Li insertion and removal kinetics in the composite material at different states of the electrode and the results suggested that Li ions experienced low impedance while charging which plausibly secured high reversible capacities. Superior specific capacity, long cycle life and better rate capability of the electrode material are due to the excellent synergy between Co2Mo3O8 nanoplatelets and rGO in the composite. These results are encouraging for the use of Co2Mo3O8/rGO composite as an anode material in the second-generation reversible Li-ion batteries.Acknowledgements
SKM and SSP are thankful to NUS for funding through India research initiative (NUS-IRI) fund (WBS No. R069000006646). SSP is grateful to Government of India for providing financial support through MANF (F1-17.1/2011/MANF-MUS-AND-213/(SA-III/Website)) to pursue research studies at the University of Hyderabad (UoH). MVR and SA thanks National Research Foundation, Prime Minister's Office, Singapore for support under its Competitive Research Program (CRP Award No. NRF-CRP 10-2012-6). Authors wish to thank Mr Henche Kuan, Department of Materials Science and Engineering, NUS, for recording XPS spectra. Authors are thankful to Center for Nanotechnology, UoH for providing TEM facility.References
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS PubMed.
- J. H. Ku, J. H. Ryu, S. H. Kim, O. H. Han and S. M. Oh, Adv. Funct. Mater., 2012, 22, 3658–3664 CrossRef CAS.
- T. Tao, A. M. Glushenkov, C. Zhang, H. Zhang, D. Zhou, Z. Guo, H. K. Liu, Q. Chen, H. Hu and Y. Chen, J. Mater. Chem., 2011, 21, 9350–9355 RSC.
- J. Kong, C. Zhao, Y. Wei, S. L. Phua, Y. Dong and X. Lu, J. Mater. Chem. A, 2014, 2, 15191–15199 CAS.
- Y. Shi, C. Hua, B. Li, X. Fang, C. Yao, Y. Zhang, Y.-S. Hu, Z. Wang, L. Chen, D. Zhao and G. D. Stucky, Adv. Funct. Mater., 2013, 23, 1832–1838 CrossRef CAS.
- Z. Ju, E. Zhang, Y. Zhao, Z. Xing, Q. Zhuang, Y. Qiang and Y. Qian, Small, 2015, 11, 4753–4761 CrossRef CAS PubMed.
- C. T. Cherian, M. V. Reddy, S. C. Haur and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 918–923 CAS.
- J. H. Ahn, G. D. Park, Y. C. Kang and J.-H. Lee, Electrochim. Acta, 2015, 174, 102–110 CrossRef CAS.
- N. Sharma, K. M. Shaju, G. V. Subba Rao, B. V. R. Chowdari, Z. L. Dong and T. J. White, Chem. Mater., 2004, 16, 504–512 CrossRef CAS.
- L.-Q. Mai, F. Yang, Y.-L. Zhao, X. Xu, L. Xu and Y.-Z. Luo, Nat. Commun., 2011, 2, 381 CrossRef PubMed.
- N. N. Leyzerovich, K. G. Bramnik, T. Buhrmester, H. Ehrenberg and H. Fuess, J. Power Sources, 2004, 127, 76–84 CrossRef CAS.
- J. Haetge, C. Suchomski and T. Brezesinski, Small, 2013, 9, 2541–2544 CrossRef CAS PubMed.
- B. Das, M. V. Reddy, C. Krishnamoorthi, S. Tripathy, R. Mahendiran, G. V. Subba Rao and B. V. R. Chowdari, Electrochim. Acta, 2009, 54, 3360–3373 CrossRef CAS.
- B. Das, M. V. Reddy, S. Tripathy and B. V. R. Chowdari, RSC Adv., 2014, 4, 33883–33889 RSC.
- J. H. Ku, Y. S. Jung, K. T. Lee, C. H. Kim and S. M. Oh, J. Electrochem. Soc., 2009, 156, A688–A693 CrossRef CAS.
- B. Guan, W. Sun and Y. Wang, Electrochim. Acta, 2016, 190, 354–359 CrossRef CAS.
- X. Xu, H. Tan, K. Xi, S. Ding, D. Yu, S. Cheng, G. Yang, X. Peng, A. Fakeeh and R. V. Kumar, Carbon, 2015, 84, 491–499 CrossRef CAS.
- S. Petnikota, K. W. Teo, L. Chen, A. Sim, S. K. Marka, M. V. Reddy, V. V. S. S. Srikanth, S. Adams and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2016, 8, 10884–10896 CAS.
- S. K. Marka and V. V. S. S. Srikanth, Nanosci. Nanotechnol.--Asia, 2015, 5, 90–108 CAS.
- D. Applestone, S. Yoon and A. Manthiram, J. Phys. Chem. C, 2011, 115, 18909–18915 CAS.
- X. Chen, H. Zhu, Y.-C. Chen, Y. Shang, A. Cao, L. Hu and G. W. Rubloff, ACS Nano, 2012, 6, 7948–7955 CrossRef CAS PubMed.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2009, 9, 72–75 CrossRef CAS PubMed.
- H. Wang, L.-F. Cui, Y. Yang, H. Sanchez Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS PubMed.
- H.-W. Shim, I.-S. Cho, K. S. Hong, A.-H. Lim and D.-W. Kim, J. Phys. Chem. C, 2011, 115, 16228–16233 CAS.
- W. Li, Y.-X. Yin, S. Xin, W.-G. Song and Y.-G. Guo, Energy Environ. Sci., 2012, 5, 8007–8013 CAS.
- W. Xiao, J. S. Chen, C. M. Li, R. Xu and X. W. Lou, Chem. Mater., 2010, 22, 746–754 CrossRef CAS.
- Y. Sun, X. Hu, W. Luo and Y. Huang, J. Mater. Chem., 2011, 21, 17229–17235 RSC.
- Y. Sun, X. Hu, W. Luo, J. Shu and Y. Huang, J. Mater. Chem. A, 2013, 1, 4468–4474 CAS.
- S. Petnikota, S. K. Marka, V. V. S. S. Srikanth, M. V. Reddy and B. V. R. Chowdari, Electrochim. Acta, 2015, 178, 699–708 CrossRef CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- S. L. Tey, M. V. Reddy, G. V. Subba Rao, B. V. R. Chowdari, J. B. Yi, J. Ding and J. J. Vittal, Chem. Mater., 2006, 18, 1587–1594 CrossRef CAS.
- K. Saravanan, J. J. Vittal, M. V. Reddy, B. V. R. Chowdari and P. Balaya, J. Solid State Electrochem., 2010, 14, 1755–1760 CrossRef CAS.
- M. Nagarathinam, K. Saravanan, E. J. H. Phua, M. V. Reddy, B. V. R. Chowdari and J. J. Vittal, Angew. Chem., Int. Ed., 2012, 51, 5866–5870 CrossRef CAS PubMed.
- A. S. Hameed, M. Nagarathinam, M. Schreyer, M. V. Reddy, B. V. R. Chowdari and J. J. Vittal, J. Mater. Chem. A, 2013, 1, 5721–5726 Search PubMed.
- A. Shahul Hameed, M. Nagarathinam, M. V. Reddy, B. V. R. Chowdari and J. J. Vittal, J. Mater. Chem., 2012, 22, 7206–7213 RSC.
- M. V. Reddy, B. L. W. Wen, K. P. Loh and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 7777–7785 CAS.
- M. V. Reddy, B. C. Zhang, K. P. Loh and B. V. R. Chowdari, CrystEngComm, 2013, 15, 3568–3574 RSC.
- B. Das, M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Mater. Chem., 2012, 22, 17505–17510 RSC.
- C. T. Cherian, M. V. Reddy, T. Magdaleno, C. H. Sow, K. V. Ramanujachary, G. V. Subba Rao and B. V. R. Chowdari, CrystEngComm, 2012, 14, 978–986 RSC.
- A. S. Hameed, H. Bahiraei, M. V. Reddy, M. Z. Shoushtari, J. J. Vittal, C. K. Ong and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2014, 6, 10744–10753 CAS.
- Y. Wu, P. Zhu, M. V. Reddy, B. V. R. Chowdari and S. Ramakrishna, ACS Appl. Mater. Interfaces, 2014, 6, 1951–1958 CAS.
- S. Petnikota, N. K. Rotte, V. V. S. S. Srikanth, B. S. R. Kota, M. V. Reddy, K. P. Loh and B. V. R. Chowdari, J. Solid State Electrochem., 2014, 18, 941–949 CrossRef CAS.
- K. S. Tan, M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2005, 141, 129–142 CrossRef CAS.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Electrochim. Acta, 2005, 50, 3375–3382 CrossRef CAS.
- M. V. Reddy, B. Pecquenard, P. Vinatier and A. Levasseur, Electrochem. Commun., 2007, 9, 409–415 CrossRef CAS.
- A. V. Murugan, M. V. Reddy, G. Campet and K. Vijayamohanan, J. Electroanal. Chem., 2007, 603, 287–296 CrossRef.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- J. Zhao, Z. Liu, Y. Qin and W. Hu, CrystEngComm, 2014, 16, 2001–2008 RSC.
- L. Soriano, M. Abbate, A. Fernández, A. R. González-Elipe, F. Sirotti and J. M. Sanz, J. Phys. Chem. B, 1999, 103, 6676–6679 CrossRef CAS.
- W. Ji, R. Shen, R. Yang, G. Yu, X. Guo, L. Peng and W. Ding, J. Mater. Chem. A, 2014, 2, 699–704 CAS.
- L.-N. Zhou, X.-T. Zhang, W.-J. Shen, S.-G. Sun and Y.-J. Li, RSC Adv., 2015, 5, 46017–46025 RSC.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
- M. V. Reddy, B. L. Wei Wen, K. P. Loh and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 7777–7785 CAS.
- M. V. Reddy, C. Y. Quan, K. W. Teo, L. J. Ho and B. V. R. Chowdari, J. Phys. Chem. C, 2015, 119, 4709–4718 CAS.
- M. V. Reddy, C. Yu, F. Jiahuan, K. P. Loh and B. V. R. Chowdari, ACS Appl. Mater. Interfaces, 2013, 5, 4361–4366 CAS.
- J. Xia, L. X. Song, W. Liu, Y. Teng, L. Zhao, Q. S. Wang and M. M. Ruan, Dalton Trans., 2015, 44, 13450–13454 RSC.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, J. Power Sources, 2010, 195, 5768–5774 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10192e |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2016 |