
Molecular relaxation and dynamic rheology of “cluster phase”-free ionomers based on lanthanum(iii)-neutralized low-carboxylated poly(methyl methacrylate)†
Abstract
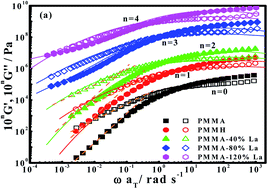
Molecular dynamics and linear dynamic rheology of La(III)-neutralized low-carboxylated poly(methyl methacrylate) (PMMA) ionomers with varying neutralization levels are investigated. While the ionomers do not form a clear cluster phase, increasing neutralization level causes notable retardation of the α relaxation and elevation of glass transition temperature. In addition, dynamic rheology of the ionomer melt follows the time–temperature superposition principle and, at neutralization levels above 80%, shows a long-term relaxation process and nonterminal relaxation ascribed to ionic species. Especially the ionomer with a neutralization level of 120% behaves like a critical gel. The long-term relaxation process is well described in terms of Cole–Cole curves, relaxation time spectra, complex viscosity and loss tangent. By analyzing the linear rheology in the framework of a “two phase” model, an interconnected multiplets network is identified as a mechanism being responsible for the fluid-to-solid transition of “cluster phase”-free ionomers with increasing neutralization level.
 Please wait while we load your content...
Please wait while we load your content...

