

Abstract
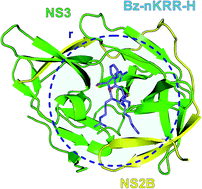
We present an in silico study of the interaction energy between NS2B–NS3, a serine protease of the dengue virus (DENV), and the inhibitor benzoyl-norleucine-Lys-Arg-Arg-aldehyde (Bz-nKRR-H), a crucial step in the design and development of dengue's antiviral drugs, using quantum chemistry calculations based on the density functional theory (DFT) at the generalized gradient approximation (GGA). The interaction energies between the inhibitor Bz-nKRR-H and each amino acid belonging to the binding site was calculated through the molecular fragmentation with conjugate caps (MFCC) approach employing a dispersion corrected exchange-correlation functional. Besides the interaction energy, we also calculated the distances, types of molecular interactions, and the atomic groups involved in the process. Our results show that the interaction energy of the system reached convergence at 15.0 Å, with the central residues identified in this interaction radius, as well as their attraction/repulsion energies, all of them being important inputs to improve the effectiveness of antiviral drugs to avoid the dissemination of the dengue virus.
 Please wait while we load your content...
Please wait while we load your content...

