
Preparation and characterization of solid lipid nanoparticles loaded with cytarabine via a micellar composition for leukemia
Abstract
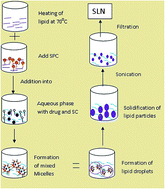
Cytarabine is an anticancer drug used in hematological malignancies and lymphoma but its short biological half life demands continuous intravenous infusion or time spaced injection. This would lead to patient incompliances and discomfort. Therefore it is imperative to look for novel therapeutic systems with lesser side effects urgently to address the underlying causes of poor treatment outcomes associated with conventional therapy. Hence entrapping of cytarabine in a solid lipid nanoparticle (SLN) based formulation could be a better alternative to sustain release and improve therapeutic activity by maintaining the plasma level. We chose tristearin as solid lipid and formulated it in nanoparticulate form by an ultrasonic emulsification method. SLN formulations were optimized and characterized for various physiochemical parameters like size (below 200 nm), charge, morphology and entrapment efficiency. An in vitro drug release study through a dialysis bag revealed a prolonged release of cytarabine. A cell line study on HL60 cell showed a significantly higher efficacy of cytarabine SLN as compared to cytarabine solution. Well characterized carriers were further subjected to hemolytic toxicity. Results showed no hemolytic toxicity and good stability at ambient temperature.
 Please wait while we load your content...
Please wait while we load your content...

