
Thermally stable ferrocene-α-cyanostilbenes as efficient materials for second order nonlinear optical polarizability†
Abstract
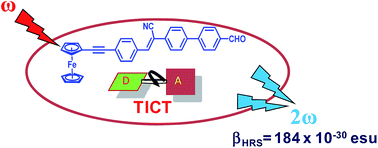
A set of new ferrocene-α-cyanostilbene (Fc-α-CNS) conjugated dyads was synthesized and their optical, nonlinear optical (NLO) and electrochemical properties were investigated. The second-order nonlinear polarizabilities were determined using hyper-Rayleigh scattering (HRS) with femtosecond pulsed laser light at 840 nm. The dyads exhibit structure-dependent NLO properties, which could be rationalized by correlating optical with electrochemical data. The dyad with the strongest (formyl) acceptor group in combination with the longer conjugation path between this acceptor and the Fc (ferrocene) donor, showed the largest static hyperpolarizability value, as compared to dyads with shorter conjugation and/or weaker acceptors. A partially symmetrical and longer triad showed the longest absorption wavelength, yet the smallest dipolar properties (dipole moment and first hyperpolarizability), are due to partial cancellation, in agreement with symmetry correlations.
 Please wait while we load your content...
Please wait while we load your content...

