
Ir(iii)-catalyzed synthesis of isoquinolines from benzimidates and α-diazocarbonyl compounds†
Abstract
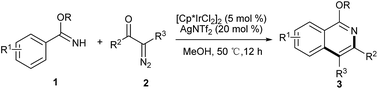
We report herein a tandem Ir(III)-catalyzed C–H activation and annulation reaction for the synthesis of isoquinolines by using readily available substituted benzimidates and α-diazocarbonyl compounds under mild conditions. The catalytic reaction exhibits excellent tolerance to different functional groups and the corresponding isoquinolines were obtained in good to excellent yields. This novel method affords an alternative strategy for the construction of diverse and useful isoquinoline derivatives.
- This article is part of the themed collection: Editors’ collection: Catalytic Organic Transformations
 Please wait while we load your content...
Please wait while we load your content...

