
Microwave-assisted methylation of dihydroxybenzene derivatives with dimethyl carbonate†
Matthew Y. Lui a,
Kapil S. Lokareb,
Ellen Hemminga,
Jessica N. G. Stanleyc,
Alvise Perosad,
Maurizio Selvad,
Anthony F. Mastersa and
Thomas Maschmeyer*ae
a,
Kapil S. Lokareb,
Ellen Hemminga,
Jessica N. G. Stanleyc,
Alvise Perosad,
Maurizio Selvad,
Anthony F. Mastersa and
Thomas Maschmeyer*ae
aLaboratory of Advanced Catalysis for Sustainability, School of Chemistry F11, The University of Sydney, Sydney, 2006, Australia. E-mail: thomas.maschmeyer@sydney.edu.au; Tel: +61 2 93512581
bHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Str. 2, 12489 Berlin, Germany
cSchool of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
dDepartment of Molecular Sciences and Nanosystems, Centre for Sustainable Chemical Technologies, Università Ca'Foscari, Venezia, Italy
eAustralian Institute for Nanoscale Science and Technology, The University of Sydney, NSW 2006, Australia
First published on 10th June 2016
Abstract
Using a focused microwave reactor, methylation with dimethyl carbonate (DMC) of 1,2- and 1,4-dihydroxybenzene derivatives, found in the product spectrum of lignin depolymerisation, leads to the respective aromatic bis-methyl ethers with excellent isolated yields. Stoichiometric as well as catalytic amounts of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) are effective for the bis-methylation of these dihydroxybenzenes at relatively mild temperatures (160–190 °C). Conversion of resorcinol (1,3-dihydroxybenzene) under similar conditions leads to a mixture of 1,3-dimethoxybenzene and methyl 2,4-dimethoxybenzoate. The unusual reactivity of resorcinol's phenyl ring towards DMC can be explained by the synergic effect of its two strongly activating ortho/para directing groups.
Introduction
Dimethyl carbonate (DMC) has been described as a green reagent and solvent.1,2 It can be prepared as a non-toxic compound via phosgene-free routes, including catalytic oxidative carbonylation reactions and CO2 cycloaddition to epoxides combined with transesterification.3–5 DMC exhibits a versatile reactivity, which makes it a candidate for the replacement of highly noxious chemicals such as phosgene, dimethyl sulfate (DMS) or methyl iodide (MeI) in catalytic methoxycarbonylation and methylation protocols1,6 (Scheme 1). | ||
| Scheme 1 Catalytic methoxycarbonylation and methylation reaction of a generic nucleophile YH with DMC (top and bottom, respectively). | ||
A detailed comparison of model methylations of O-, C- and N-nucleophiles has proven that DMC-mediated reactions are not only safer and more atom economic, but they are also advantageous in terms of mass index with respect to processes using DMS or MeI.7 DMC may also be preferred over methanol for alkylation reactions: in this case, the choice is mainly dictated by other features, particularly the non-toxicity of DMC and the relatively lower alkylating temperatures (160–250 °C for DMC versus >320 °C for MeOH).7
The technique of microwave heating has become a valuable method for the synthesis of organic compounds. Traditional heating techniques (e.g. oil baths on hot plates) are not always the most effective method to transfer energy to the reactants of a reaction mixture, especially since they depend on the thermal conductivity of the various materials that must be permeated.8 Conversely, with microwave heating, molecules in a reaction mixture might be efficiently heated internally by the direct coupling of energy from microwave irradiation. Furthermore, modern microwave instruments allow for precise temperature control,8 as well as enabling reaction mixtures to be superheated in a closed vessel. The latter feature is particularly useful for alkylation processes with DMC, as it allows the operating temperatures for these reactions to be considerably higher than DMC's boiling point (90 °C), reducing the reaction time. It is however accepted by the majority of scientists that a non-thermal microwave effect does not exist.9
In the specific case of O-methylation reactions, most of the DMC-based procedures use simple alcohols and phenols as substrates.10 Both conventional and microwave heating techniques have been employed for the methylation of phenols. Tundo et al. reported the conversion of simple phenols with DMC in the presence of K2CO3 under reflux conditions (90 °C) for 53 hours.11 The conversion rate was very slow and in most cases a mixture of methylation and carboxymethylation products was formed. Lee et al. described the methylation of phenol with DMC in the presence of Cs2CO3 in a sealed tube at 120 °C.10b Using this method, 1-naphthol was completely methylated after 4 hours of heating. On the other hand, Glasnov et al. reported the microwave-assisted methylation of 4-nitrophenol and 4-fluorophenol with DMC using tributylmethylammonium methyl carbonate, which could be generated in situ with tributylamine, as the catalyst.10k The process was efficient and gave high yields of the O-methylation products under flow microwave conditions. The procedure, however, required the use of high temperatures and pressures (285 °C and 150 bar), which could be detrimental to operational safety. On the other hand, only a few examples have been described for the methylation of dihydroxybenzenes (DHBs) with DMC. These compounds (DHBs) and the corresponding ethers are of major interest as intermediates for cosmetics and pharmaceuticals. Moreover, studies on the synthetic potential of DHBs and their derivatives are further fueled by the fact that such aromatics are building blocks of the lignin scaffold. In fact, lignin depolymerisation mainly affords catechol,12 but also several other DHBs including hydroquinone,13,14 resorcinol,15 4-methylcatechol,16–21 4-ethylcatechol,17,18 methylhydroquinone16,22 and dimethylhydroquinone.23,24 It is noteworthy that the corresponding methyl ethers of all these compounds find application across a remarkable range as solvents and synthetic intermediates for the production of fragrances, cosmetics, pharmaceuticals and fuel additives.25
The O-alkylation of DHBs with DMC currently requires very high temperatures. For example, the continuous-flow alkylation of both catechol and hydroquinone with DMC was reported over supported KOH catalysts at 280 °C:26 under such conditions, mixtures of mono- and bis-O-methyl derivatives are observed. Ono and co-workers also reported the reaction of catechol with DMC in the gas phase at 320 °C, yielding guaiacol or veratrole (mono- and bis-methyl ether products, respectively) in the presence of CsOH/Al2O3 or KX/Al2O3 [X = NO3−, HCO3−, (CO3−)(1/2)] as catalysts.27d,e The same process was also investigated over MgO-based catalysts between 250 and 320 °C, where a ∼70% selectivity towards guaiacol was achieved.27i In general, other procedures always afforded mixtures of mono- and bis-O-methyl products, with the occasional formation of mono-C-methyl derivatives.27
Alternative methods for the alkylation of DHBs may be more selective towards the corresponding bis-methyl ethers, but they resort to the use of hazardous reagents such as MeI,28 DMS,29 and methanol,30 or even the explosive N,N-dimethylformamide–dimethylacetal (DMF–DMA).31 In this context, the design of a safe and efficient strategy for the syntheses of such ethers is a highly desirable target and it becomes even more attractive from the perspective of using not only biomass-derived aromatics, but also mild reaction conditions to optimise the product distribution. As part of a program on green methodologies for the upgrading of renewables, we have recently approached this problem by reporting on the reaction of DMC with 4-(3-hydroxypropyl)phenol, in the presence of several inorganic- and organo-catalysts including K2CO3, CsF/α-Al2O3, alkali metal-exchanged faujasites, and trioctylmethylphosphonium methyl carbonate ([P8881][CH3OCOO]), respectively.32 Although a good alkylation selectivity (>80%) was obtained, the reaction took place only at T ≥ 180 °C, requiring the use of steel autoclaves under an autogenous pressure of 6–10 bar.
Here, we describe that the O-methylation of three models of DHBs such as catechol, resorcinol, and hydroquinone, has been achieved with dimethyl carbonate by using a focused microwave system under liquid-phase conditions. This is a largely unexplored area with only a very few papers describing microwave-assisted methylation of just the simple (mono)-phenols by DMC using 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a catalyst in MeCN, DMF, or sulfolane solvents.10d–g
This work elucidates paths towards not only a significant decrease of reaction times (down to minutes from the hours for conventionally heated processes), but it also improves selectivity, safety and facilitates a more convenient separation of products – all working together to enhance the ‘green’ character of this type of conversion.
Materials and methods
General
All reactions were carried out in air. DMC, mesitylene, catechol, 4-methylcatechol, 4-ethylcatechol, methylhydroquinone, dimethylhydroquinone, DBU, DABCO and Cs2CO3 (Aldrich) resorcinol, pyridine, K2CO3 and KOH (Ajax), triethylamine (Merck) hydroquinone (Alfa Aesar) and catechol carbonate (Oxchem) were used without purification.Quantification of the reaction products was performed on a Shimadzu GC-2010 Plus gas chromatograph equipped with Rtx-5MS 0.25 μm × 30 m × 0.25 mm column under the following conditions. The temperature program had an isothermal period of 1 min at 50 °C, then the temperature was increased by 15 °C min−1 to another isothermal period of 4 min at 250 °C. Compounds of interest were quantified using calibration curves of authentic samples. In some cases, compounds were quantified by 1H NMR analysis. NMR spectra were collected using a Bruker 300 MHz instrument at about 300 K after 16 scans. 1H NMR spectra were obtained using a 90° flip angle with a recycle delay (D1) of 100 seconds. Compound identification by GC-MS was performed on a Shimadzu GCMS-QP2010 equipped with Rtx-5MS 0.25 μm × 30 m × 0.25 mm column under the following conditions. The temperature program had an isothermal period of 3 min at 50 °C. Then the temperature was increased by 10 °C min−1 to another isothermal period of 10 min at 330 °C. Compounds were identified by comparing the EI-MS spectrum with the MS library NIST05. The Anton Paar Monowave 300 microwave synthesis reactor used in the experiments below has the following specifications, magnetron frequency, 2455 MHz; power consumption, 1600 VA; and an AC 230 V ± 10% 50 Hz/60 Hz power supply.
Determination of vapor pressure of DMC
A batch reactor (245 mL internal volume, Parr Instrument Company, modified with the inclusion of a second external thermocouple) was charged with DMC (60 mL, 0.71 mol). The reactor was purged with N2 and pressurised to 6760 mbar with N2. The reactor was heated stepwise in integral control mode under continuous stirring, holding the temperature at each of the chosen set-points (below) for 10 min, to allow for equilibration. The heating steps were: 22.6 °C – hold – 80 °C – hold – 140 °C – hold – 160 °C – hold – 180 °C – hold – 220 °C – hold –230 °C – hold – 240 °C – hold. The reactor pressure was recorded approximately every 10 min. The measurement series was performed twice and the data sets combined.Methylation of catechol, hydroquinone and resorcinol with DMC at various temperatures under microwave conditions
A solution of substrate (2.73 mmol), DBU (5.46 mmol), DMC (3 mL), and MeCN (3 mL) was heated in a 30 mL air tight glass vessel in an Anton Paar Monowave 300 microwave synthesis reactor at temperature “T” °C with a stirring rate of 600 rpm. After 40 minutes, the reaction vial was removed from the microwave reactor and cooled to room temperature. The mixture was concentrated at 40 °C and 98 mbar, and mesitylene (200 μL) added as an external standard. The mixture was then combined with 1 M HCl (7.5 mL) and magnesium chloride hexahydrate (7.7 M, 7.5 mL). The organic layer was subsequently extracted with ethyl acetate (10 × 2 mL), dried over anhydrous sodium sulfate and filtered. The sample was then analysed by 1H NMR, GC-FID and GC-MS.Methylation of catechol with various amounts of DMC under microwave conditions
A solution of catechol (2.73 mmol), DBU (5.46 mmol), MeCN (3 mL) and various amount of DMC was heated in a 30 mL air tight glass vessel in an Anton Paar Monowave 300 microwave synthesis reactor at temperature of 160 °C with a stirring rate of 600 rpm. After 40 minutes, the reaction vial was removed from the microwave reactor and cooled to room temperature. The mixture was concentrated at 40 °C and 98 mbar, and mesitylene (200 μL) added as an external standard. The mixture was then combined with 1 M HCl (7.5 mL) and magnesium chloride hexahydrate (7.7 M, 7.5 mL). The organic layer was subsequently extracted with ethyl acetate (10 × 2 mL), dried over anhydrous sodium sulfate and filtered. The sample was then analysed by GC-FID and GC-MS.Methylation of catechol, hydroquinone and resorcinol with DMC using catalytic amounts of DBU under microwave conditions
A solution of substrate (2.73 mmol), a catalytic amount of DBU, DMC (3 mL) and MeCN (3 mL) was heated in a 30 mL air tight glass vessel in an Anton Paar Monowave 300 microwave synthesis reactor at different temperatures with a stirring rate of 600 rpm. After 80 minutes, the reaction vial was removed from the microwave reactor and cooled to room temperature. The mixture was concentrated at 40 °C and 98 mbar, and mesitylene (200 μL) added as an external standard. The mixture was then combined with 1 M HCl (7.5 mL) and magnesium chloride hexahydrate (7.7 M, 7.5 mL). The organic layer was subsequently extracted with ethyl acetate (10 × 2 mL), dried over anhydrous sodium sulfate and filtered. The sample was then analysed by 1H NMR, GC-FID and GC-MS.Methylation of various dihydroxybenzenes using the focused microwave
A glass tube (30 mL) was loaded with a stirrer bar, substrate (2.73 mmol), DBU (5.46 mmol), DMC (3 mL) MeCN (3 mL). The tube was then fitted with a Teflon cap, and heated at the assigned temperature for the requisite time in an Anton Paar Monowave 300 microwave synthesis reactor with a stirring rate of 600 rpm. After heating was completed, the pressure tube was removed from the microwave reactor and cooled to room temperature, and the volatiles were removed in vacuo. The mixture was then combined with 1 M HCl (7.5 mL) and magnesium chloride hexahydrate (7.7 M, 7.5 mL). The organic layer was subsequently extracted with ethyl acetate (3 × 10 mL). The organic fractions were combined and concentrated. The product mixture was purified by column chromatography on silica gel. The chromatography was carried out using mixtures of hexane/diethyl ether (from 100% hexane to 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v hexane/diethyl ether) as eluents.
:3 v/v hexane/diethyl ether) as eluents.
Methylation of catechol carbonate with DMC under microwave conditions
A solution of catechol carbonate (0.91 mmol), various amounts of DBU and methanol, DMC (1 mL), and MeCN (1 mL) was heated in a 10 mL air tight glass vessel in an Anton Paar Monowave 300 microwave synthesis reactor at 160 °C with a stirring rate of 600 rpm. The reaction vial was removed from the microwave reactor and cooled to room temperature. The mixture was concentrated at 40 °C and 98 mbar, and mesitylene (100 μL) added as an external standard. The mixture was then combined with 1 M HCl (7.5 mL) and magnesium chloride hexahydrate (7.7 M, 7.5 mL). The organic layer was subsequently extracted with ethyl acetate (10 × 2 mL), dried over anhydrous sodium sulfate and filtered. The sample was then analysed by GC-FID and GC-MS.Synthesis of methyl 4-hydroxy-2-methoxybenzoate (compound 6)
Methyl 4-hydroxy-2-methoxybenzoate was synthesised following a procedure33 reported previously.Results and discussion
Methylation reactions involving DMC are typically performed at temperatures ≥160 °C, i.e. much higher than its boiling point (90 °C). The knowledge of the vapour pressure of DMC is therefore important for the design and safe operation of such reactions. However, few of such data are available in the literature, referring only to a narrow temperature range below 155 °C.34As the first part of this investigation, measurements of the vapour pressure of DMC were carried out from ambient to high temperatures under isochoric conditions: in particular, the total pressure in a batch reactor containing N2 and DMC was determined across the range of 22.6 to 240 °C (295.75 to 513.15 K).
The vapour pressures of DMC as a function of varying temperature were fitted to the integrated form of the Clausius–Clapeyron equation (eqn (1)):
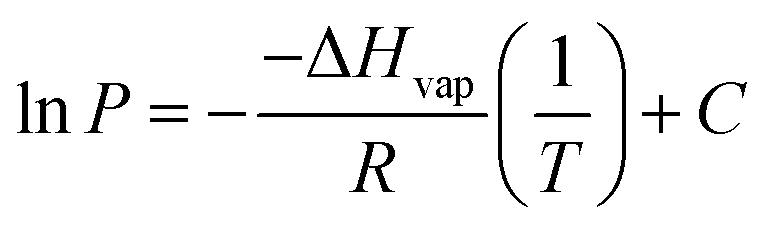 | (1) |
Eqn (1) assumes that the enthalpy of vapourisation is constant across the entire temperature range. Furthermore, DMC is assumed to behave as an ideal gas. The fit of the data indicates sufficient linearity to render these assumptions valid for the purposes discussed here (cf. Fig. 1 the plot of ln(P/kPa) vs. 103 T/K−1 with a straight fit and an R-squared value of 0.91).
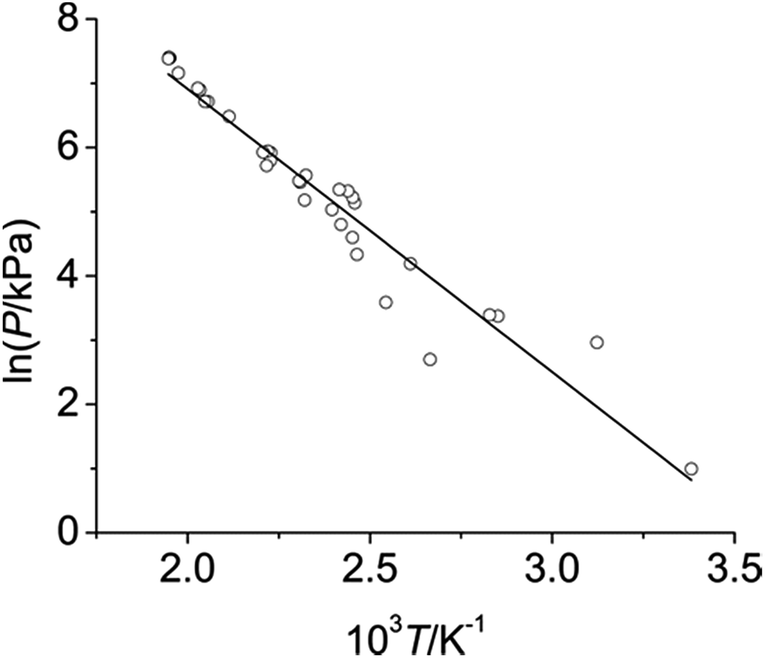 | ||
| Fig. 1 Graph of ln(P/kPa) versus 103 T/K−1, combining data sets from the two measurements. Raw data shown as open circles, fitted data using eqn (1), see above, shown as solid line. | ||
To verify the reliability of the measurements obtained, and the appropriateness of using the Clausius–Clapeyron equation for the system investigated, the enthalpy of vapourisation over the experimental temperature range was determined from the fit as 36.6 ± 3.4 kJ mol−1 (2σ error). This is in excellent agreement with those reported in the literature by both Steele et al. (37.7 kJ mol−1 at 25 °C)35 and Walker (37.3 kJ mol−1 at 25 °C).36
This result confirmed that DMC was stable under the conditions examined. Although the data were not obtained under perfect equilibrium conditions, the excellent fit with the literature values reported for lower temperature ranges, suggests that the approximation to equilibrium conditions is acceptable. These results can serve as an empirically based guide to the planning and assessment of reactions of DMC across an extended temperature range – something that is currently absent in the literature.
Phenol as substrate
To rank our system's performance, initially we investigated the well-established methylation of phenol with DMC, which was carried out in a focused microwave as a screening test for a number of common, commercially available bases. Of all the bases tested, DBU afforded almost complete conversion of phenol within 20 min (93.2%, Table S1, ESI†) in MeCN as determined by GC-FID analysis (mesitylene as an internal standard). The exceptional activity of DBU shown here is consistent with the earlier reports by Tilstam under continuous flow,10f,g and Rajabi utilizing a conventional microwave oven.10d Other bases examined include 1,4-diazabicyclo[2.2.2]octane (DABCO), triethylamine, pyridine, KOH, K2CO3 and Cs2CO3 (Table S1, ESI†). While DABCO afforded 85.3% yield, triethylamine afforded only a moderate yield, 14.1%, and pyridine gave just 0.4% yield. Additionally, KOH, K2CO3 and Cs2CO3 afforded 4.7, 6.2 and 63.7% yields respectively. The more soluble Cs2CO3 affords a significantly higher conversion as compared to less soluble bases such as KOH and K2CO3 in MeCN.Attention was then switched to the less/unexplored DHBs such as catechol, hydroquinone and resorcinol.
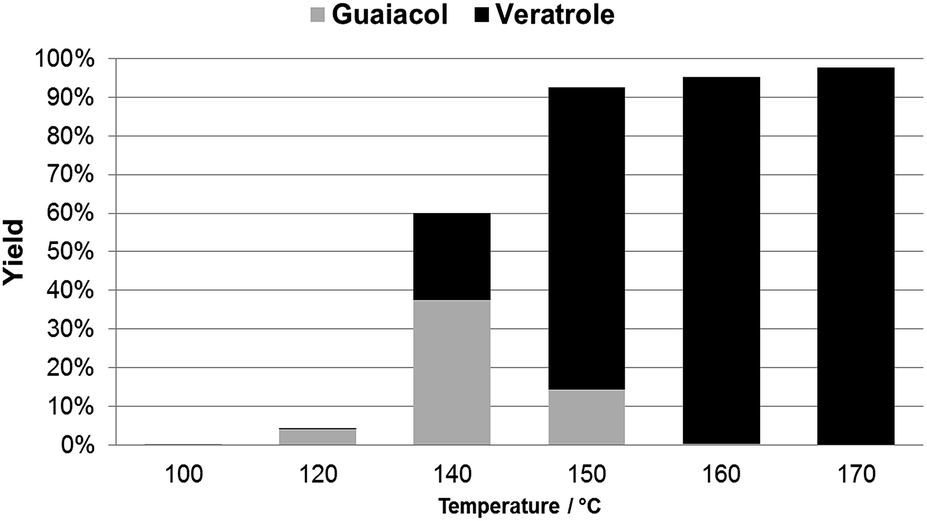 | ||
| Fig. 2 Yield profile for mono- and bis-methylation of catechol (2.73 mmol) with dimethyl carbonate (3 mL) in the presence of DBU (5.46 mmol) and CH3CN (3 mL). Reactions were carried out for 40 min, in an Anton Paar 300 focused microwave, at T of 100–170 °C. Yield was determined by GC. | ||
Beside catechol, guaiacol and veratrole, traces of catechol carbonate (<1%) were observed in some of the reaction mixtures. In particular, at 160 °C, catechol carbonate was detected only in the early stage of the reaction. Although this compound has been reported as one of the intermediates in the conversion of catechol to veratrole, no mechanism has yet been suggested for this transformation.10d,e,j
To explore this aspect, pure catechol carbonate was set to react under conditions similar to those used for catechol (with and without methanol). Excellent yields of veratrole were obtained (Table 1). Recognising that an equilibrium between DBU, DMC and the methoxide ion is established (Fig. 3, frame window, top),38,39 a plausible mechanism for the formation of veratrole is illustrated at the bottom of Fig. 3: an initial transesterification promoted by CH3O− is followed by intramolecular decarboxylation producing the anion (ii); this is then methylated either by DMC or the N-methoxycarbonylated salt of DBU (i) (paths a and b, respectively).
| Entry | Catechol carbonate/mmol | Methanol/mmol | Time/min | Guaiacol/% | Veratrole/% |
|---|---|---|---|---|---|
| 1 | 0.91 | 0 | 16 | 16.4 | 41.7 |
| 2 | 0.91 | 0 | 32 | 2.5 | 90.1 |
| 3 | 0.91 | 0 | 64 | 0 | 93.0 |
| 4 | 0.18 | 0 | 64 | 0 | 91.6 |
| 5 | 0.91 | 1.82 | 64 | 0 | 94.2 |
| 6 | 0.91 | 9.1 | 128 | 0 | 89.5 |
 | ||
| Fig. 3 One possible mechanism for the formation of veratrole from catechol carbonate (ring opened by methoxide). | ||
Alternatively, DBU could also undergo direct nucleophilic attack on the catechol carbonate (Fig. 4). This opens up the carbonate ring and frees a phenoxide ion for reaction with the methylating agent (i.e. the N-methoxycarbonyl-amidinium adduct in Fig. 3). Then transesterification of the resulting intermediate by the methoxide ion releases free DBU and generates 2-methoxyphenyl methyl carbonate, which reacts with DBU to form the N-methoxycarbonyl-amidinium cation and guaiacyl anion. Subsequently, the two ionic species react to produce veratrole.
 | ||
| Fig. 4 Alternative mechanism for the formation of veratrole from catechol carbonate (ring-opened by DBU). | ||
The methylation of catechol was then investigated in the presence of other bases. Results followed a trend similar to that observed for phenol (Table 2): at 170 °C, Cs2CO3 was not as active as DBU (compare entries 2, 3 and 4); while the use of DABCO resulted in the full conversion of catechol after 40 minutes, it was poorly selective due to the formation of unidentified by-products (entry 5).
| Entry | Substrate | Base | Temp/°C | Conv'n/% | Mono-methylated product yield/% | Bis-methylated product yield/% |
|---|---|---|---|---|---|---|
| a Heated for 80 min. | ||||||
| 1 | Catechol | DBU | 160 | 100 | 0.3 | 94.9 |
| 2 | Catechol | DBU | 170 | 100 | 0 | 97.6 |
| 3 | Catechol | Cs2CO3 | 170 | 100 | 23.0 | 57.4 |
| 4a | Catechol | Cs2CO3 | 170 | 100 | 8.1 | 70.3 |
| 5 | Catechol | DABCO | 170 | 100 | 0 | 70.4 |
| 6 | Hydroquinone | DBU | 160 | 96.3 | 19.6 | 71.9 |
| 7 | Hydroquinone | DBU | 170 | 100 | 0 | 96.1 |
| 8 | Resorcinol | DBU | 170 | 100 | 0 | 35.0 |
| 9 | 0.5 Catechol + 0.5 hydroquinone | DBU | 170 | 100 | 0 | 88.2 + 83.7 |
 | ||
| Fig. 5 Pathways of the reaction of resorcinol with DBU and DMC. | ||
Of note, the methylation of 4 afforded only compound 5, but not even a trace of the isomer methyl 4-hydroxy-2-methoxybenzoate (6, Fig. 5) was observed in the reaction mixtures, indicating that the hydroxy substituent para to the methyl ester group was more sterically accessible, than that in the ortho position. Then, only once 5 was formed, the subsequent methylation step took place, forming methyl 2,4-dimethoxybenzoate (7, Fig. 5). No carboxymethylation of the phenyl rings was observed when either 3-methoxyphenol, 2, or 1,3-dimethoxybenzene, 3, was heated under the same conditions, indicating that ring carboxymethylation, rather than oxo-methylation, of the resorcinolate dianion is preferred. When no DBU was added, resorcinol did not react. Hence, oxoanions, which are more activating than hydroxy groups,40,41 must be created for the carboxymethylation to occur.
After the electron-withdrawing methyl ester group was introduced to the phenyl ring, the phenyl ring is no longer nucleophilic enough40 to react with a second DMC molecule, making methyl 2,4-dimethoxybenzoate the only major side-product of this process.
Effects of reaction conditions
| Entry | DMC/catechol ratio | Guaiacol/% | Veratrole/% |
|---|---|---|---|
| 1 | 2.2 | 34.0 | 6.1 |
| 2 | 4.4 | 47.6 | 28.4 |
| 3 | 8.7 | 0.8 | 94.8 |
| 4 | 13.1 | 0.3 | 94.9 |
| 5 | 17.5 | 0 | 90.3 |
 | ||
| Fig. 6 The reaction of resorcinol (2.73 mmol) with DMC (3 mL) in the presence of different amounts of DBU. Experiments were carried out at 190 °C with CH3CN (3 mL) as a solvent. Mixtures were heated for 10 min except for the reaction using a DBU/–OH ratio of 0.0625 for which heating was prolonged up to 80 min. Yield was determined by 1H NMR (connecting lines as visual guide only). | ||
All reaction mixtures were heated until the conversions of resorcinol and compounds 2, 4 and 5 were complete. The increase of the DBU/1 ratio from 2 (one base equiv. per each OH group of 1) to 16, favoured the formation of the undesired product methyl 2,4-dimethoxybenzoate (7), the amount of which was progressively raised from 55 to 75%. In all of these cases, compound 7 was the major product. By contrast, if only 6.25 mol% of DBU was used, the O-methylation selectivity improved significantly and 1,3-dimethoxybenzene (3) could be obtained in a yield as high as 84%. This not only demonstrated that the relative amounts of DBU and resorcinol affected the product distribution of the reaction, but also that the base could be used in a catalytic amount.
An explanation for the observed variation in selectivity is offered by considering the properties of oxoanions of resorcinol. It should be noted that in water, at 25 °C, the pKa values of resorcinol and the resorcinolate anion almost overlap (9.20 and 10.90).42 Therefore, the corresponding mono-deprotonated and bis-deprotonated species are likely to co-exist. Under non-aqueous conditions (as those used in this work), it is reasonable to assume that a similar equilibrium (but with a different equilibrium constant) exists among the mono- and di-anion of resorcinol. If so, when a high DBU/substrate ratio is used, most of the resorcinol is bis-deprotonated. However, in the presence of a catalytic amount of DBU (6.25 mol%), the mono oxoanion of resorcinol is almost exclusive formed. This species alone does not sufficiently activate the phenyl ring towards C-carboxylation, thereby hindering the formation of methyl 2,4-dimethoxybenzoate (7). As mentioned above, only in the bis-oxoanion of resorcinol, synergistic ortho/para directing/activating effects may operate to impart an unusually high reactivity of the 4- and 6-positions of the aromatic ring.
Other substrates
The reaction conditions optimised for the methylation of phenol, catechol, hydroquinone and resorcinol, were then extended to the study of the O-methylation of several other DHBs as potential derivatives of the depolymerisation of the primary lignin network.12–24 In particular, the reaction of 4-methylcatechol, 4-ethylcatechol, methylhydroquinone and 2,3-dimethylhydroquinone with DMC were investigated at 160–170 °C with MW-heating and in the presence of DBU (Table 4). All substrates tested were converted into the corresponding bis-O-methylated products with excellent isolated yields (85–93%), thus confirming the general efficacy of the proposed method.DBU as a catalyst
Fig. 6 clearly demonstrated that DBU could be used in catalytic amounts with respect to resorcinol to achieve the corresponding O-bis-methyl derivative. These results perfectly match the general behavior of all DMC-mediated methylation reactions that are genuine catalytic processes (Scheme 1). To further investigate this aspect and fully prove the potential of the proposed protocol, additional experiments were carried out on the reaction of catechol and hydroquinone with DMC by using catalytic amounts of DBU. Results are reported in Table 5. For both catechol and hydroquinone, the decrease of the substrate/DBU molar ratio to 4 or 8 (12.5% and 6.25% molar equivalents of catalyst with respect to –OH groups, respectively) still facilitated the formation of the corresponding bis-methyl ethers in high yields (87–92%, entries 3, 4, and 5). At the same time however, the temperature had to be increased from 160 to 180 and 190 °C to complete the reaction.| Entry | Substrate | Substrate/DBU (mol/mol) | Temp/°C | Mono-methylated product/% | Bis-methylated product/% |
|---|---|---|---|---|---|
| 1 | Catechol | 60 | 170 | 49.5 | 1.8 |
| 2 | Catechol | 60 | 180 | 0 | 92.4 |
| 3 | Catechol | 60 | 180 | 33.9 | 0.7 |
| 4 | Catechol | 40 | 190 | 0 | 90.1 |
| 5 | Hydroquinone | 60 | 190 | 0 | 87.2 |
Shieh et al. suggested that DBU is not simply a base but also a nucleophilic catalyst which reacts with DMC to produce a more activated methylating agent (Fig. 3).10e Once the alkylation is complete, the methoxide ions generated can act as proton scavengers to liberate the protonated DBU and allow the reaction to proceed further.10e However, the methoxide ion itself may also act as a nucleophilic activator by converting the substrate into the corresponding oxoanion(s).
Conclusions
A method for the bis-O-methylation of a series of dihydroxybenzene derivatives has been developed by using dimethyl carbonate as a green reagent in a focused microwave reactor. Excellent isolated yields (up to 93%) have been achieved for the corresponding ether products. Overall, the procedure represents an efficient approach for the upgrading of lignin building blocks, especially 1,2-dihydroxybenzene and 1,4-dihydroxybenzene derivatives, that can be selectively converted under relatively mild conditions with DBU as an inexpensive catalyst.Additional studies directed at expanding the scope of this transformation, for example the conversion of dihydroxybenzenes with higher alkyl carbonates, the methylation of mixtures of such derivatives generated by HTU of lignocellulosic biomass, and the application of a flow microwave reactor for these processes will be the subject of future investigations.
Acknowledgements
The authors thank the Science and Industry Endowment Fund (SIEF) (Grant no. RP03-028) for partial financial support for this research, JNGS acknowledges the receipt of an Australian Postgraduate Award, and we thank Dr Alex Yuen for valuable discussions.References
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706 CrossRef CAS PubMed.
- B. Schaffner, F. Schaffner, S. P. Verevkin and A. Borner, Chem. Rev., 2010, 110, 4554 CrossRef CAS PubMed.
- U. Romano, F. Rivetti and N. Di Muzio, Anic S.p.A., US Pat., US4318862(A), 1982.
- K. Nishhira, K. Mizutare and S. Tanaka, UBE Industries, Japan, US Pat., US5162563(A), 1992.
- S. Fukuoka, I. Fukawa, M. Tojo, K. Oonishi, H. Hachiya, M. Aminaka, K. Hasegawa and K. Komiya, Catal. Surv. Asia, 2010, 14, 146 CrossRef CAS.
- (a) M. Selva, P. Tundo, A. Perosa and F. Dall'Acqua, J. Org. Chem., 2005, 70, 2771 CrossRef CAS PubMed; (b) M. Selva, Pure Appl. Chem., 2007, 79(11), 1855 CrossRef CAS.
- M. Selva and A. Perosa, Green Chem., 2008, 10, 457 RSC.
- (a) C. O. Kappe, Chem. Soc. Rev., 2008, 37, 1127 RSC; (b) C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250 CrossRef CAS PubMed; (c) C. O. Kappe and D. Dallinger, Chem. Rev., 2007, 107, 2563 CrossRef PubMed.
- (a) N. Kuhnert, Angew. Chem., Int. Ed., 2002, 41, 1863 CrossRef CAS; (b) M. A. Herrero, J. M. Kremsner and C. O. Kappe, J. Org. Chem., 2008, 73, 36 CrossRef CAS PubMed; (c) C. O. Kappe, B. Pieber and D. Dallinger, Angew. Chem., Int. Ed., 2013, 52, 1088 CrossRef CAS PubMed.
- (a) G. Barcelo, D. Grenouillat, J.-P. Senet and G. Sennyey, Tetrahedron, 1990, 46, 1839 CrossRef CAS; (b) Y. Lee and I. Shimizu, Synlett, 1998, 1063 CrossRef CAS; (c) S. Ouk, S. Thiébaud, E. Borredon and P. Le Garrs, Green Chem., 2002, 4, 431 RSC; (d) F. Rajabi and M. R. Saidi, Synth. Commun., 2004, 34, 4179 CrossRef CAS; (e) W. Shieh, S. Dell and O. Repič, Org. Lett., 2001, 3(26), 4279 CrossRef CAS PubMed; (f) U. Tilstam, Org. Process Res. Dev., 2012, 16, 1150 CrossRef CAS; (g) U. Tilstam, Org. Process Res. Dev., 2012, 16, 1974 CrossRef CAS; (h) P. Tundo, F. Trotta, G. Moraglio and F. Ligorati, Ind. Eng. Chem. Res., 1988, 27, 1565 CrossRef CAS; (i) A. Bomben, M. Selva and P. Tundo, Ind. Eng. Chem. Res., 1999, 38, 2075 CrossRef CAS; (j) M. Selva, E. Militello and M. Fabris, Green Chem., 2008, 10, 73 RSC; (k) T. N. Glasnov, J. D. Holbrey, C. L. Kappe, K. R. Seddon and T. Yan, Green Chem., 2012, 14, 3071 RSC.
- P. Tundo, L. Rossi and A. Loris, J. Org. Chem., 2005, 70, 2219 CrossRef CAS PubMed.
- (a) R. Beauchet, F. Monteil-Rivera and J. M. Lavoie, Bioresour. Technol., 2012, 121, 328 CrossRef CAS PubMed; (b) H. Pińkowska and P. Wolak, Fuel, 2013, 113, 340 CrossRef; (c) A. Toledano, L. Serrano and J. Labidi, Fuel, 2014, 116, 617 CrossRef CAS; (d) R. Katahira, A. Mittal, K. McKinney, X. Chen, M. P. Tucker, D. K. Johnson and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 1474 CrossRef CAS.
- M. Garcia-Perez, S. Wang, J. Shen, M. Rhodes, W. J. Lee and C.-Z. Li, Energy Fuels, 2008, 22, 2022 CrossRef CAS.
- S. I. Yang, M. S. Woo and C. Y. Woo, Energy, 2014, 66, 162 CrossRef CAS.
- N. Chatterjee, H.-J. Eom, S.-H. Jung, J.-S. Kim and J. Choi, Environ. Toxicol., 2014, 29, 1409 CrossRef CAS PubMed.
- G. Yildiza, F. Ronssea, R. Venderboschb, R. van Durenc, S. R. A. Kerstend and W. Prinsa, Appl. Catal., B, 2015, 168–169, 203 CrossRef.
- K. Sipilaè, E. Kuoppala, L. Fagernaès and A. Oasmaa, Biomass Bioenergy, 1998, 14, 103 CrossRef.
- Y. Yu, Y. Zeng, J. Zuo, F. Ma, X. Yang, X. Zhang and Y. Wang, Bioresour. Technol., 2013, 134, 198 CrossRef CAS PubMed.
- H. Zhang, Y.-T. Cheng, T. P. Vispute, R. Xiao and G. W. Huber, Energy Environ. Sci., 2011, 4, 2297 CAS.
- S. Karagöz, T. Bhaskar, A. Muto and Y. Sakata, Bioresour. Technol., 2006, 97, 90 CrossRef PubMed.
- R. D. Barrows and D. C. Elliott, Fuel, 1984, 63, 4 CrossRef CAS.
- A. Aho, N. Kumar, A. V. Nashkul, K. Eränen, M. Ziolek, P. Decyk, T. Salmi, B. Holmbom, M. Hupa and D. Yu Murzin, Fuel, 2010, 89, 1992 CrossRef CAS.
- S. Karagöz, T. Bhaskar, A. Muto, Y. Sakata, T. Oshiki and T. Kishimoto, Chem. Eng. J., 2005, 108, 127 CrossRef.
- S. Karagöz, T. Bhaskar, A. Muto and Y. Sakata, Fuel, 2004, 83, 2293 CrossRef.
- E. Fuhrmann and J. Talbiersky, Org. Process Res. Dev., 2005, 9, 206 CrossRef CAS.
- P. Tundo, in Continuous-flow Methods in Organic Synthesis, Ellis Horwood Ltd, Chichester (UK), 1st edn, 1996 Search PubMed.
- (a) S. Ratton, Rhone-Poulenc Industries, US Pat., 4487975, 1984; (b) Y. Fu, T. Baba and Y. Ono, Appl. Catal., A, 1998, 166, 419 CrossRef CAS; (c) Y. Fu, T. Baba and Y. Ono, Appl. Catal., A, 1998, 166, 425 CrossRef CAS; (d) Y. Fu, T. Baba and Y. Ono, Appl. Catal., A, 1999, 178, 219 CrossRef CAS; (e) Y. Fu, T. Baba and Y. Ono, Appl. Catal., A, 1999, 176, 201 CrossRef CAS; (f) T. M. Jyothi, T. Raja, M. B. Talawar, K. Sreekumar, S. Sugunan and B. S. Rao, Synth. Commun., 2000, 30(21), 3929 CrossRef CAS; (g) T. M. Jyothi, T. Raja, M. B. Talawar and B. S. Rao, Appl. Catal., A, 2001, 211, 41 CrossRef CAS; (h) Z. L. Shen, X. Z. Jiang, W. M. Mo, B. X. Hu and N. Sun, Green Chem., 2005, 7, 97 RSC; (i) M. Vijayaraj and C. S. Gopinath, J. Catal., 2006, 243, 376 CrossRef CAS; (j) A. Dhakshinamoorthy, A. Sharmila and K. Pitchumani, Chem.–Eur. J., 2010, 16, 1128 CrossRef CAS PubMed.
- (a) A. Loupy, J. Sansoulet and F. Vaziri-Zand, Bull. Soc. Chim. Fr., 1987, 1027 CAS; (b) J. C. Lee, J. Y. Yuk and S. H. Choi, Synth. Commun., 1995, 25, 1367 CrossRef CAS; (c) P. M. Paduraru, R. T. W. Popoff, R. Nair, R. Gries, G. Gries and E. Plettner, J. Comb. Chem., 2008, 10, 123 CrossRef CAS PubMed; (d) J. J. V. Eynde and I. Mailleux, Synth. Commun., 2001, 37, 1 CrossRef; (e) B. Behrmand, F. Molin and H. Gallardo, Dyes Pigm., 2012, 95, 600 CrossRef; (f) P. J. Reddy, A. S. Reddy, J. S. Yadav and B. V. S. Reddy, Tetrahedron Lett., 2012, 53, 4051 CrossRef; (g) V. Karunakaran, D. D. Prabhu and S. Das, J. Phys. Chem. C, 2013, 117, 9404 CrossRef CAS.
- (a) W. H. Perkin Jr and C. Weizmann, J. Chem. Soc., Trans., 1906, 89, 1649 RSC; (b) E. H. Vickery, L. F. Pahler and E. J. Eisenbraun, J. Org. Chem., 1979, 44(24), 4444 CrossRef CAS; (c) J. Zhang, Y. Cui, M. Wang and J. Liu, Chem. Commun., 2002, 2526 RSC; (d) A. R. Massah, M. Mosharafian, A. R. Momeni, H. Aliyan, H. J. Naghash and M. Adibnejad, Synth. Commun., 2007, 37, 1807 CrossRef CAS; (e) S. O. Mihigo, W. Mammo, M. Bezabih and K. Andrae-Marobela, Bioorg. Med. Chem., 2010, 18, 2464 CrossRef CAS PubMed; (f) U. S. Hiremath, Tetrahedron Lett., 2013, 54, 3419 CrossRef CAS; (g) H. Singh, A. Balamurugan and M. Jayakannan, ACS Appl. Mater. Interfaces, 2013, 5, 5578 CrossRef CAS PubMed.
- (a) K. Bauer, R. Molleken and R. Hopp, Haarmann & Reimer GmbH, US Pat., US3895076(A), 1975; (b) G. T. McCloud, M. R. Brimer and C. L. Gibson, Eastman Kodak Company, US Pat., US3911022(A), 1975; (c) S. Porchet, S. Su, R. Doepper and A. Renken, Chem. Eng. Technol., 1994, 17, 108 CrossRef CAS; (d) S. C. Lee, S. W. Lee, K. S. Kim, T. J. Lee, D. H. Kim and J. C. Kim, Catal. Today, 1998, 44, 253 CrossRef CAS; (e) V. Vishwanathan, S. Ndou, L. Sikhwivhilu, N. Plint, K. V. Raghavan and N. J. Coville, Chem. Commun., 2001, 893 RSC; (f) R. Bal, S. Mayadevi and S. Sivasanker, Org. Process Res. Dev., 2003, 7, 17 CrossRef CAS; (g) X. Zhu, X. Li, M. Jia, G. Liu, W. Zhang and D. Jiang, Appl. Catal., A, 2005, 282, 155 CrossRef CAS; (h) X. Zhu, X. Li, X. Zou, Y. Wang, M. Jia and W. Zhang, Catal. Commun., 2006, 7, 579 CrossRef CAS; (i) X. Liao, Z. Zhou, Z. Wang, X. Zou, G. Liu, M. Jia and W. Zhang, J. Colloid Interface Sci., 2007, 308, 176 CrossRef CAS PubMed; (j) A. A. Jafari, A. Khodadadi and Y. Mortazavi, J. Mol. Catal. A: Chem., 2013, 372, 79 CrossRef CAS.
- P. Belov, V. L. Campanella, A. W. Smith and R. Priefer, Tetrahedron Lett., 2011, 52, 2776 CrossRef CAS.
- J. N. G. Stanley, M. Selva, A. F. Masters, T. Maschmeyer and A. Perosa, Green Chem., 2013, 15, 3195 RSC.
- D. W. Robertson, E. E. Beedle, J. H. Krushinski, G. D. Pollock, H. Wilson, V. L. Wyss and J. S. Hayes, J. Med. Chem., 1985, 28, 717 CrossRef CAS PubMed.
- Y. Zhou, J. Wu and E. W. Lemmon, J. Phys. Chem. Ref. Data, 2011, 40(4), 043106 CrossRef.
- W. V. Steele, R. D. Chirico, S. E. Knipmeyer, A. Nguyen and N. K. Smith, J. Chem. Eng. Data, 1997, 42(6), 1037 CrossRef CAS.
- E. E. Walker, J. Appl. Chem., 1952, 2, 470 CrossRef CAS.
- W. H. Walker, A. R. Collett and C. L. Lazzell, J. Phys. Chem., 1931, 35, 3259 CrossRef CAS.
- M. Carafa, E. Mesto and E. Quaranta, Eur. J. Org. Chem., 2011, 2458 CrossRef CAS.
- Y. Ji, J. Sweeney, J. Zoglio and D. Gorin, J. Org. Chem., 2013, 78, 11606 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- R. F. W. Bader and C. Chang, J. Phys. Chem., 1989, 93, 2946 CrossRef CAS.
- S. E. Blanco, M. C. Almandoz and F. H. Ferretti, Spectrochim. Acta, Part A, 2005, 61, 93 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra09841j |
| This journal is © The Royal Society of Chemistry 2016 |