
Gold nanoparticle-mediated electron transfer of cytochrome c on a self-assembled surface†
Abstract
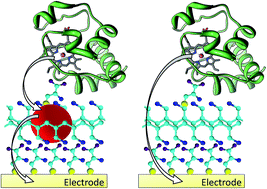
The presence of gold nanoparticles (AuNPs) at the protein/electrode interface has a significant impact on the electrodic microenvironment, and allows the optimization of the activity catalysis as well as electrochemical properties. Here, we report a novel and accurate methodology to observe AuNP mediated electron transfer mechanism from Cytochrome c (Cyt c) to a polycrystalline gold surface. Poly(allylamine hydrochloride) molecules (PAH) were used as spacers between Cyt c and the electrode surface, and the electron rate constant within the PAH layer was measured in the presence and absence of AuNPs. Based on cyclic voltammetric experiments and Marcus theory, a four-fold increase in the electron rate constant was observed in the presence of AuNPs, and the reorganization energy was estimated to be 0.49 eV. Furthermore, AuNPs decreased the effective distance between the redox center of Cyt c and the electrode surface by 20%. These results suggest that the electron transfer properties of Cyt c based protein electrodes are significantly enhanced in the presence of the AuNPs.
 Please wait while we load your content...
Please wait while we load your content...

