
New insights into the sensing mechanism of shape controlled ZnO particles†
Massimiliano D'Arienzo*a,
Matteo Redaellia,
Barbara Di Credicoa,
Stefano Polizzib,
Roberto Scottia and
Franca Morazzonia
aINSTM, Department of Materials Science, University of Milano-Bicocca, Via R. Cozzi 55, I-20125 Milano, Italy. E-mail: massimiliano.darienzo1@unimib.it
bDepartment of Molecular Sciences and Nanosystems, University Ca' Foscari of Venezia, Via Torino 155/b, I-30172 Venezia, Italy
First published on 25th May 2016
Abstract
The sensing behavior of pyramidal (PY), prismatic hexagonal (EP) and hexagonal rod-like (ER) ZnO micro and sub-microparticles, has been compared with that of commercial ZnO (ZnO®) particles having nanometric size and uneven shape. The performances have been firstly related to the predominance of specific crystal surfaces and then, more in depth, to the paramagnetic defects in ZnO ( and
and  ), detected by Electron Spin Resonance (ESR), in order to associate the particles morphology with the defects amount and reactivity and, in turn, with a particular sensing mechanism. The results showed that the sensing behavior of ZnO® containing irregular nanoparticles is essentially related to the alternate formation and filling of oxygen vacancies during the gas pulse (oxygen vacancy mechanism), while that of ER and EP crystals does not seem to directly involve the
), detected by Electron Spin Resonance (ESR), in order to associate the particles morphology with the defects amount and reactivity and, in turn, with a particular sensing mechanism. The results showed that the sensing behavior of ZnO® containing irregular nanoparticles is essentially related to the alternate formation and filling of oxygen vacancies during the gas pulse (oxygen vacancy mechanism), while that of ER and EP crystals does not seem to directly involve the  defects. In particular, the sensing properties of shape controlled ZnO particles are mainly attributed to the ability of (0001) exposed surfaces in favoring a far better chemisorption of negatively charged oxygen species, then available for the reactions with the reducing gas (i.e. ionosorption mechanism). The outcomes and the approach adopted in this study may positively contribute to the debate still existing between the oxygen vacancy and ionosorption models by giving indications on the predominance of a specific sensing mechanism in shape controlled ZnO.
defects. In particular, the sensing properties of shape controlled ZnO particles are mainly attributed to the ability of (0001) exposed surfaces in favoring a far better chemisorption of negatively charged oxygen species, then available for the reactions with the reducing gas (i.e. ionosorption mechanism). The outcomes and the approach adopted in this study may positively contribute to the debate still existing between the oxygen vacancy and ionosorption models by giving indications on the predominance of a specific sensing mechanism in shape controlled ZnO.
Introduction
Over the past decades, considerable efforts have been made to improve the electrical response and the selectivity of semiconductor metal-oxide based gas sensors, through a fine tuning of size, shape and, more recently, of the particle exposed crystal surfaces.1–6In this context, we recently reported on the relationships between the morphological features, specifically the exposed crystal surfaces of SnO2 (ref. 5) and WO3 (ref. 6) nanocrystals, and their sensing properties. In particular, in the case of shape controlled SnO2, we demonstrated, on the basis of comprehensive electron spin resonance (ESR) investigation, that different faces could differently stabilize the oxygen defects (in form of singly ionized oxygen vacancies,  ). The
). The  amount increases under reducing gas treatment, while it decreases in the presence of oxidative treatments. Thus, the electrical modifications of shape controlled SnO2 under gas pulse were attributed to alternate formation and filling of oxygen vacancies, confirming the oxygen vacancy mechanism already suggested and validated by us in sol–gel obtained SnO2 (ref. 5 and 7–9) and by Barsan et al. in colloidal SnO2 through in operando DRIFT spectroscopy.13 This mechanism is alternative to the ionosorption mechanism, which instead attributes the conductivity variations to chemisorption and removal of oxygen anions located at the oxide surface.10–12,14,15
amount increases under reducing gas treatment, while it decreases in the presence of oxidative treatments. Thus, the electrical modifications of shape controlled SnO2 under gas pulse were attributed to alternate formation and filling of oxygen vacancies, confirming the oxygen vacancy mechanism already suggested and validated by us in sol–gel obtained SnO2 (ref. 5 and 7–9) and by Barsan et al. in colloidal SnO2 through in operando DRIFT spectroscopy.13 This mechanism is alternative to the ionosorption mechanism, which instead attributes the conductivity variations to chemisorption and removal of oxygen anions located at the oxide surface.10–12,14,15
Along the above directions, we have explored the studies concerning the relationships between morphology and sensing properties in ZnO, one of the most promising oxide materials for gas sensor applications.13–15
In this context, Kuang et al.16 have recently described the sensitivity to ethanol of ZnO micro/nano crystals with flake, rod-like and pyramidal shape, containing different percentages of (0001) (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0), (101) and (000) surfaces. They found that nanoflakes, which highly expose the (0001) facets, exhibit gas-sensing performance towards ethanol better than the particles with predominant (100), (101) and (000) facets. The differences in the electrical response were attributed to the different ability of the exposed facets in chemisorbing oxygen. In fact since the (0001) face is terminated by Zn2+ ions, the chemisorption ability of this face is very high, because the surface cations easily seize oxygen anionic species through physical/chemical absorption.
0), (101) and (000) surfaces. They found that nanoflakes, which highly expose the (0001) facets, exhibit gas-sensing performance towards ethanol better than the particles with predominant (100), (101) and (000) facets. The differences in the electrical response were attributed to the different ability of the exposed facets in chemisorbing oxygen. In fact since the (0001) face is terminated by Zn2+ ions, the chemisorption ability of this face is very high, because the surface cations easily seize oxygen anionic species through physical/chemical absorption.
Another recent investigation,17 reported on ZnO nanorods array with highly exposed (0001) facets, which exhibit enhanced sensing properties at T > 573 K. This behavior was attributed both to the higher surface energy of the (0001) facets compared to the (100) and (101) ones, and to the presence of undercoordinated cations, i.e. three and two-fold coordinated Zn2+. Thus, the (0001) surface contains more oxygen defects than the other ones, which can significantly promote the chemisorption of oxygen anionic species.
Both these studies hypothesize the occurrence of the ionosorption mechanism,10–12,14,15 even if no spectroscopic evidences were supplied to support either the predominance of this mechanism or the possible involvement of a different one.
In our previous studies on commercial ZnO nanoparticles (ZnO®),18–20 we invoked the role of paramagnetic defects in the sensing mechanism, namely  and interstitial zinc
and interstitial zinc  , suggesting a vacancy based mechanism. Nevertheless the connections among defects, size/shape of the particles and the sensing mechanism were not investigated.
, suggesting a vacancy based mechanism. Nevertheless the connections among defects, size/shape of the particles and the sensing mechanism were not investigated.
Trying to elucidate the sensing mechanism in shape controlled ZnO, micro and sub-microcrystals with pyramidal (PY), prismatic hexagonal (EP) and hexagonal rod-like (ER) shapes, having specific exposed crystal surfaces, were synthesized by a tailored solvothermal route in the presence of oleic acid (OA) and oleyl amine (OM) as surfactants. These molecules have been already proved to be highly effective in driving the growth of the crystals along specific crystallographic directions, allowing to obtain different morphologies depending on both the metal precursor/OA/OM ratio and the reaction temperature.21
The sensing behaviors of PY, EP and ER samples toward CO were examined and compared to that of commercial ZnO® nanoparticles with uneven size and shape.
Moreover, the obtained performances were discussed in connection with the paramagnetic defects detected by electron spin resonance (ESR), in order to associate the type, the amount and the reactivity of these defects to the morphology of the particles and in turn, to the sensing mechanism.
It resulted that, while the sensing ability of ZnO® nanoparticles is essentially related to an oxygen vacancy mechanism,5,7–9 for shape controlled ZnO crystals the oxygen ionosorption mechanism seems to prevail.10–12
The overall results and the adopted approach may positively contribute to the debate still existing between the oxygen vacancy and ionosorption models for the sensing of semiconductor metal-oxides.
Experimental section
Chemicals
Zinc acetate (Zn(OAc)2 or ZA, 97%), oleic acid (C18H33CO2H or OA, 90%), oleylamine (C18H35NH2 or OM, 70%) were all purchased from Aldrich and used without further purification. Nanocrystalline commercial ZnO was purchased from USP-1 from the Zinc Corporation of America (ZnO®).Synthesis of shape controlled ZnO crystals
Solvothermal synthesis of shape controlled ZnO micro and nanocrystals was performed by reacting the ZA in the presence of OA and OM. In a typical experiment ZA (6.25 mmol) was added to a mixture of X mmol OA, Y mmol OM, and 5.76 g of absolute ethanol. X and Y were varied to gain different ZA/OA/OM molar ratios, which led to differently shaped microcrystals: pyramidal (PY, ZA/OA/OM = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:6), hexagonal prismatic (EP, ZA/OA/OM = 1:8:2), and hexagonal rods (ER, ZA/OA/OM = 1:0:8).
:4:6), hexagonal prismatic (EP, ZA/OA/OM = 1:8:2), and hexagonal rods (ER, ZA/OA/OM = 1:0:8).
For example, in order to synthesize PY ZnO, 6.25 mmol of ZA were added to 25 mmol of OA, 37.5 mmol of OM, and 5.76 g of absolute ethanol. The obtained mixture was stirred for 15 minutes, then transferred into a 400 mL Teflon-lined stainless steel autoclave containing 25 mL of absolute ethanol 96%. The mixture was then heated to 140 °C or 200 °C for 20 h. After decantation, ZnO powder was recovered from the autoclave, washed several times with ethanol in order to completely remove the residual amounts of organic surfactants, then filtered and finally dried under vacuum (p < 10−2 mbar) at room temperature (RT).
The scheme of the solvothermal synthesis of ZnO particles is reported in Fig. 1. Geometrical models showing the surfaces exposed by PY and ER, EP ZnO crystals are also given.
 | ||
| Fig. 1 Guide lines to obtain shape controlled ZnO crystals. Right side: Geometrical model of pyramidal ZnO (PY) exposing six {101} and one (000) facets, of hexagonal prismatic ZnO (EP) and hexagonal rods ZnO (ER) with {100}, (0001) and (000) exposed surfaces. | ||
Structural, morphological and spectroscopic characterization
X-ray diffraction (XRD) patterns of tin oxide nanocrystals were collected with a Bruker D8 Advance (Cu Kα radiation) in the range 20–70° 2θ (2θ step 0.025°, count time of 2 s per step).Scanning electron microscopy (SEM) measurements of the PY, ER and EP particles were performed by a Vega TS5136 XM Tescan microscope in a high-vacuum configuration. The electron beam excitation was 30 kV at a beam current of 25 pA, and the working distance was 12 mm. In this configuration the beam spot was 38 nm. Prior to SEM analysis samples were gold-sputtered.
High-resolution transmission electron microscopy (HR-TEM) was performed on ZnO commercial nanoparticles using a Jeol 3010 apparatus operated at 300 kV with a high-resolution pole piece (0.17 nm point-to-point resolution) and equipped with a Gatan slow-scan 794 CCD camera. Samples were obtained by removing a film portion from the substrates in order to obtain a fine powder sample, then suspended in 2-propanol. A 5 μL drop of this suspension was deposited on a holey carbon film supported on a 3 mm copper grid for TEM investigation.
In order to verify the complete removal of the residual surfactants after purification, Attenuated Total Reflection – Fourier transform infrared spectroscopy (ATR-FTIR) analysis on shape controlled ZnO particles was performed by a Perkin Elmer Spectrum 100 instrument (4 cm−1 resolution spectra, 650–4000 cm−1 region, 32 scans). To further evaluate the removal of these organic molecules, Thermo Gravimetric Analysis (TGA) was also performed. TGA curves were collected by a Mettler Toledo TGA/DSC1 STARe System, at constant gas flow (50 cm3 min−1). The sample powders were heated in air from 30 to 1000 °C at heating rate of 10 °C min−1. Thermal profile was the following: 40 °C for 5 min (under N2); 40–150 °C 10 °C min−1 (under N2); 150 °C for 5 min (under N2); 150–1000 °C 10 °C min−1 (under air).
XPS was also used to assess the presence of C contaminants on the surface of the samples. Spectra were obtained by a Perkin-Elmer Φ 5600-ci spectrometer using non-monochromatized Al Kα radiation (1486.6 eV). Samples were mounted on steel holders and introduced directly into the fast-entry lock system of the XPS analytical chamber. The analysis area was 800 μm in diameter and the working pressure was lower than 10−9 mbar. The spectrometer was calibrated by assuming the binding energy (BE) of the Au4f7/2 line at 83.9 eV with respect to the Fermi level. The standard deviation for the BEs values was ±0.2 eV. Survey scans were obtained in the 0–1300 eV range. Detailed scans were recorded for the C1s, O1s, Na1s, Si2p and Zn2p regions. No further element was detected. Charging effects were corrected by assigning to the C1s peak associated with adventitious hydrocarbons a value of 284.8 eV.22 The analysis involved Shirley-type background subtraction23 non-linear least-squares curve fitting adopting Gaussian–Lorentzian peak shapes, and peak area determination by integration.
Nitrogen physisorption experiments were performed by using a Quantachrome Autosorb-1 apparatus. Specific surface area (SSABET) by the BET method24 and pore size distribution of mesopores by BJH method,25 were determined only for ER and ZnO® samples, since these methods are appropriate for particles with sub-micrometric or nanometric size. The powders were evacuated at 200 °C for 16 h before the analysis. Instead, for PY and EP microcrystals, the surface area was calculated by measuring the average dimensions (l and h) of 100 particles from SEM images and by using the density of ZnO (d = 5.606 g cm−3). The derived surface area was labeled as SSAGEO. In order to confirm this procedure, the SSAGEO was calculated also for ER particles and the obtained value was compared to the SSABET.
In detail, the SSAGEO of PY particles was calculated considering as model an PY with side of the hexagonal basis l (Fig. 1, right side), by the following equation:
 | (1) |
The model for EP and ER particles is approximately that of a hexagonal prism with height h and side of the hexagonal basis l (Fig. 1, right side). Their SSAGEO was calculated according to the equation:
 | (2) |
By using this latter model, the percentages of the high-energy (0001) exposed crystal surfaces for ER and EP particles were calculated according to the equation:
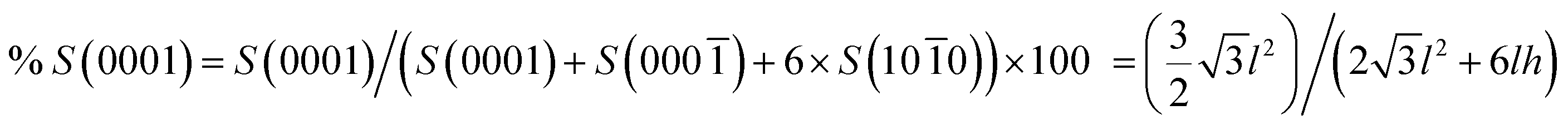 | (3) |
Electrical measurements
The films for the sensing measurements were prepared by simply depositing by drop-casting few drops of ZnO paste (consisting of ZnO particles mixed with ethanol) onto Suprasil quartz slides (20 × 20 mm, 0.25 mm thickness). Suprasil quartz slides were equipped, before film deposition, with two gold current collectors (20 mm) deposited at a distance of about 2 mm from each other by the dc sputtering technique. The thickness of the resulting sensing layer was approximately 200 μm. Then the samples were placed in a quartz chamber sited in an oven, and the measurements performed at temperatures ranging from 200 to 350 °C. The electrical resistance was measured by a programmable electrometer controlled by a PC. To dynamically reproduce environmental conditions in a controlled and reproducible way, a system based on volumetric gas mixing through mass flow controllers and certified bottles was used. The sensing element was initially equilibrated in air flow (100 mL min−1) at the selected temperature, then CO (72.5–580 ppm)/air mixture was introduced (100 mL min−1) up to equilibrium conditions. The initial resistance conditions of the film were restored by air equilibration, before again introducing the CO/air mixture. The electrical response (S) is defined as the ratio between the film resistance under flowing air, RAIR, and under flowing CO/air mixture, RMIX, respectively (S = RAIR/RMIX).In order to carry out the electrical measurements at each operating temperature, the samples were maintained in the sensing apparatus for about 3 weeks. We tested the ZnO sensors initially at the lowest working temperature (200 °C). Then the films were heated up to 350° (the highest working T) and the resistance was recorded. After that, the measurements were performed at progressively lower temperatures (from 350 to 225 °C) and again at 200 °C to check the reproducibility and the stability of the electrical response. The results evidenced that the electrical response initially detected at 200 °C and again re-measured after the heating up at higher temperatures, remains more or less constant with an uncertainty of ±15%. This suggests the absence of any memory effect affecting the performance of the materials.
The reliability as well as the reproducibility of the devices is also satisfactory; in fact, all the produced sensors (at least two for each material) were working, and the response reproducibility (obtained by at least six pulses) was calculated to be ±13%.
ESR investigation
The electron spin resonance (ESR) investigation was performed by a Bruker EMX spectrometer operating at the X-band frequency and equipped with an Oxford cryostat, working in the temperature range of 4–298 K. The EPR spectra were recorded on shape controlled ZnO crystals and on ZnO® in form of:(a) As prepared samples;
(b) Samples treated under air stream (80 cm3 min−1) for 30 min at 250 °C or at 350 °C;
(c) Samples treated under CO (580 ppm)/Ar mixture stream (80 cm3 min−1) for 30 min at 250 °C or at 350 °C; CO/Ar mixture was used as gas probe in order to better distinguish the electronic effects due to reducing gas from those due to the oxidizing one.
(d) Samples interacted with oxygen stream (80 cm3 min−1) at RT after treatment (c), for 10 min at 298 K.
The sensing materials were treated in situ (at operating temperature, under gas exposure), and the spectra recorded at 130 K after evacuation and rapid quenching of the samples to preserve the treatment effects. Spectra were recorded with a modulation frequency of 100 kHz, modulation amplitudes of 2–5 gauss, and microwave powers of 10 mW. The g values were calculated by standardization with α,α′-diphenyl-β-picrylhydrazyl (DPPH). The spin concentration was obtained by double integration of the resonance lines, referring to the area of the standard Bruker weak pitch (9.7 × 1012 ± 5% spins per cm). Accuracy on double integration was ±15%. Care was taken to always keep the most sensitive part of the ESR cavity (1 cm length) filled.
Results
Structural and morphological characterization
Fig. 2 shows the XRD patterns of the solvothermal and commercial ZnO® particles. | ||
| Fig. 2 Right-side: XRD patterns of PY, EP, ER and commercial ZnO micro or sub-microcrystals. Left-side: Magnification of the diffraction pattern of shape controlled ZnO samples in the region between 30 and 40 2θ. | ||
The diffraction patterns of all samples have been indexed based on the hexagonal crystal system with space group P63mc, typical of the wurtzite phase of bulk zinc oxide (JCPDS no. 79-2205). No additional diffraction peaks due to impurities or amorphous phases were detected (Fig. 2, right-side). A gradual increase in the relative intensity and broadening is detectable for the (0002) diffraction peak going from PY to ER particles (Fig. 2 left-side). This suggests a growth of the crystals along the [0001] direction and a decreased average particle size, respectively.
Fig. 3 summarizes the SEM investigations performed on ZnO shape controlled particles.
 | ||
| Fig. 3 SEM images at different magnifications of (a and b) ER, (c and d) PY and (e and f) EP shape controlled ZnO micro or sub-microcrystals. | ||
When the reaction is carried out at high temperature (200 °C) and in the absence of OA (Zn/OA/OM = 1:0:8), hexagonal ZnO rods with sub-micrometric size, length ranging from 500 nm to 1 μm and preferentially exposing the (0001), (000−1) and (10−10) crystal surfaces, are obtained (Fig. 1 and 3a and b).
By introducing OA at the same reaction temperature, but leaving unchanged the concentrations of both the zinc precursor and oleylamine (Zn/OA/OM = 1:2:8), crystals with different shapes, resembling that of hexagonal pyramid or pyramid terminated on the bottom with thin hexagonal prisms exposing (000−1), (10−10) and (10−11) surfaces, have been produced (Fig. 1 and S1a in ESI†). The particles present a broad size distribution with average length between 2.5 and 5.0 μm. Increasing the amount of OA (Zn/OA/OM = 1:4:6), pyramidal microcrystals with (000−1) and (10−11) exposed surfaces, highly homogeneous in size (average height 2.5 μm) and shape are observed (Fig. 1 and 3c and d).
Finally, a further increase of OA concentration and lower concentration of OM (Zn/OA/OM = 1:8:2) results in the formation of microcrystals with different morphology as a function of the reaction temperature. At 200 °C, pseudo-spherical microcrystals with highly irregular size and shape have been obtained (Fig. S1b, ESI†). Instead, at a lower reaction temperature (140 °C), ZnO grows as very uniform hexagonal microprisms with average height of 1.6 μm and (0001), (000−1) and (10−10) exposed crystal surfaces (Fig. 3e and f).
The synthesis of ZnO particles with various shapes and dimensions have been extensively explored in the literature. Very recently, Niederberger et al.26 reported that the hydrolysis of zinc oxide precursors in the presence of amines (OA, dodecylamine or hexadecylamine) in alcoholic media leads to the crystallization of hexagonal rods, probably through particle ripening processes. This is consistent with the morphology of ER sub-microcrystals (Fig. 3a and b) observed for Zn/OA/OM = 1:0:8.
They also reported that, keeping the hexagonal shape of the basal plane, but progressively suppressing the growth along the c-axis ([0001] direction) by using suitable capping molecules (e.g. oleic acid) able to bind on the polar (0001) surface, hexagonal pyramids and prisms can be obtained. This again agrees with the shape of PY and EP microcrystals, obtained in the presence of increasing amounts of oleic acid (Fig. 3c–f).
We may also suggest that, at high temperature and when a large excess of OA is used, the acid can bind on almost all of the surfaces of ZnO seeds,27 inducing the formation of pseudo-spherical microcrystals as observed for Zn/OA/OM = 1:8:2 at reaction temperature of 200 °C, (Fig. S1b, ESI†).
SEM images were also collected on commercial ZnO, but poor information about the morphological features of the particles were gained (see Fig. 4a). For this reason, ZnO® nanopowders were characterized in depth by TEM microscopy. Particles are single nanocrystals, organized in irregular aggregates (Fig. 4b) and mainly show elongated rectangular shape with the major dimension ranging from 200 to 500 nm and the minor from 45–100 nm (see higher magnifications in Fig. 4c and d). Due to the uneven morphology of the nanoparticles, it is difficult to clearly recognize the presence of specific crystallographic planes.
 | ||
| Fig. 4 SEM micrograph (a) and TEM images at increasing magnifications (b–d) of ZnO® nanoparticles. | ||
Nitrogen physisorption experiments were performed only on ER and ZnO® particles, since the BET and BJH methods are not applicable to the large PY and EP microcrystals.
In agreement with the SEM images (Fig. 3a and b), ER sub-microcrystals show a pseudo-type II Brunauer isotherm, which is typical of macroporous materials or mesoporous materials with irregular pore systems (Fig. 5a). ZnO® nanoparticles show instead a type IV isotherm with a small hysteresis loop, indicative of the presence of small mesopores (Fig. 5b). This is consistent with the corresponding pore-size distribution (inset Fig. 5b), which shows the presence of a broad peak in the range of 2–10 nm.
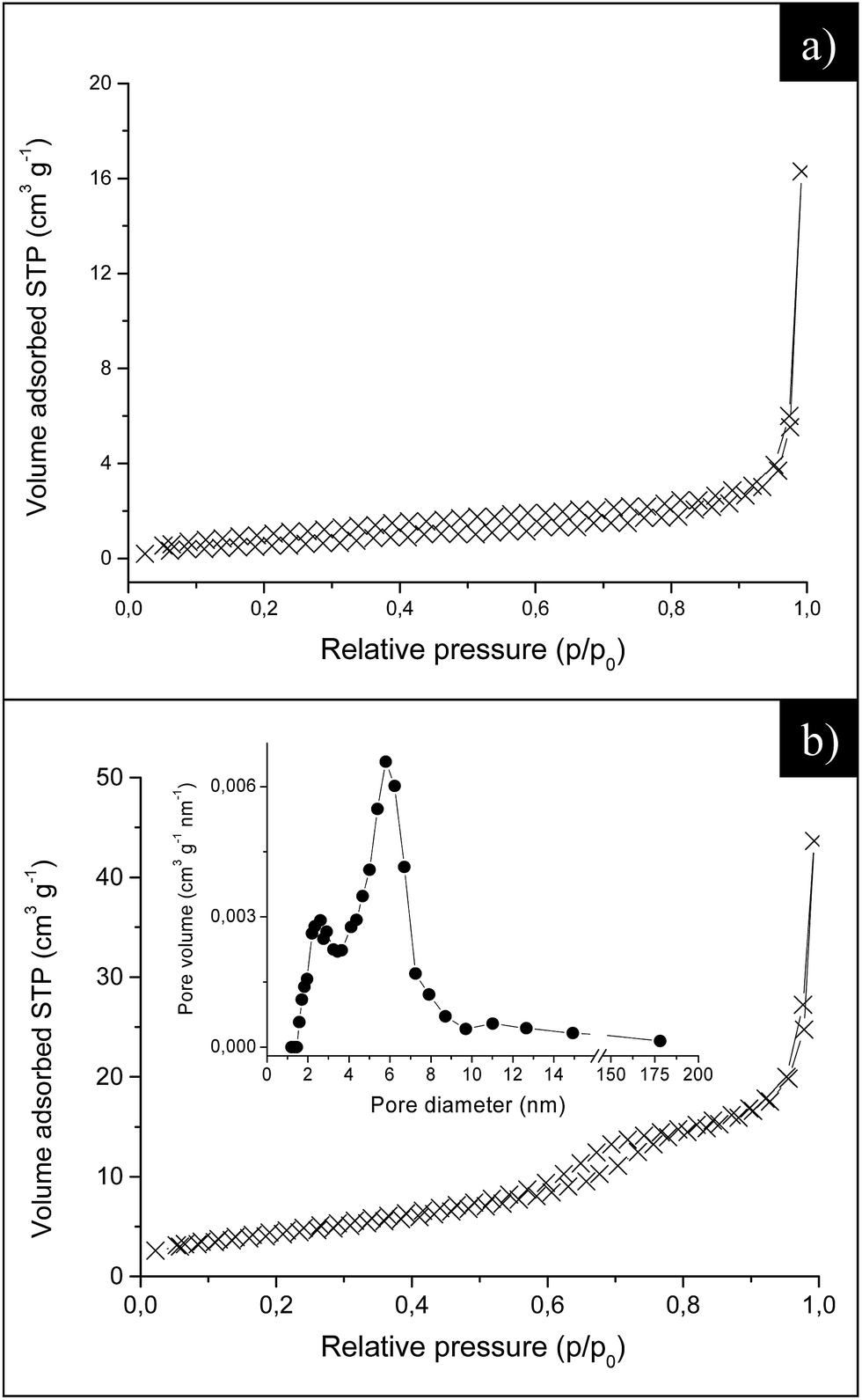 | ||
| Fig. 5 Adsorption/desorption isotherms at liquid nitrogen temperature for (a) ER and (b) ZnO® samples. The curves correspond to a pseudo-type II (a) and type IV (b) isotherms. Inset in (b) pore size distribution. | ||
The specific surface areas (SSABET) of ER and ZnO® samples are reported in Table 1. The specific surface area for PY and EP large microcrystals was instead estimated by using the simplified geometrical models described in the Experimental section and labeled as SSAGEO (see Table 1). To assess the validity of this procedure the SSAGEO has been calculated also for ER particles and compared to the specific surface area derived by nitrogen physisorption. The results show that SSAGEO and SSABET display similar values (Table 1), thus confirming the reliability of the geometrical calculations.
| Sample | SSABET (m2 g−1) | SSAGEO (m2 g−1) | Exposed (0001) crystal facets (%) | SSA of exposed (0001) crystal facets (m2 g−1) |
|---|---|---|---|---|
| ER | 4.18 | 3.85 | 25.3 | 1.06 |
| EP | — | 1.62 | 36.6 | 0.60 |
| PY | — | 0.84 | — | — |
| ZnO® | 15.9 |
In order to determine the relative surface area of the exposed (0001) surfaces by shape controlled ZnO, the percentages of (0001) were calculated. In detail, by analyzing SEM images, we measured l and h average values, which correspond to the side of the hexagonal (0001) surface and to the height of the rectangular {1010} surfaces in EP and ER crystals, respectively (see Fig. 1). These values were entered in eqn (3) (See Experimental section) to gain the percentages of (0001) exposed crystal faces and finally multiplied for the specific surface area of for EP and ER particles (Table 1). According to these calculations, we can infer that ER sub-micro particles present a surface area of (0001) faces higher than EP microcrystals.
The possible presence of surfactant traces on the surface of morphology-controlled ZnO particles, after washing with ethanol solution, was initially checked either by ATR-FTIR spectroscopy or TGA analysis. As a representative example, the IR spectrum and the thermogravimetric weight loss curve of EP microcrystals are reported in Fig. S2 (ESI†). The spectrum shows a weak band at 3370 cm−1 corresponding to water and/or hydroxyl groups, two broad and very weak bands at 1544 and 1513 cm−1 which may be related to the symmetrical and asymmetrical stretching modes of residual carboxylate groups, and a sharper peak at 953 cm−1 attributable to Zn–OH stretching.28 The corresponding TGA curve, displays a single weight loss (∼1%) between 30 and 400 °C, ascribed to the removal of the surface physisorbed cleaning solvents and of the residual carboxylate groups. Similar ATR-FTIR and TGA results were obtained for ER and PY particles.
In order to assess the nature of the small surface C contamination detected by FT-IR and TGA on shape controlled ZnO samples after the recovering and cleaning procedure, XPS analysis was also performed. Detailed scans were recorded for the C1s, O1s, and Zn2p regions. No further elements were detected. In particular, Fig. S3 (ESI†) shows the XPS survey in the C1s region for EP microcrystals, which were prepared by using the highest amount of OA capping agent (see Experimental section and Fig. 1).
The carbon amount is on the order of a few% and is mainly due to adventitious hydrocarbon contamination (C1s = 284.9 eV). However, by spectra deconvolution, a weak component associable to carbonate groups can be detected at a higher binding energy (288.3 eV).29 To verify the nature of this contribution, we tried to add in the fitting a component deriving from the double bond C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C present in the OA chain and usually detected at 284.4 eV.29 The results were inconsistent with the chemical composition of the surfactant (i.e. CC/COO = 5:1 instead of 2:1) and a significant worsening of the fitting quality was achieved. Therefore, we conclude that the component at 288.3 eV cannot be associated to remaining surfactant units but, more reasonably, derives from very small residual traces of unreacted zinc acetate precursor, which probably give rise to the weak bands observed in FT-IR spectra and decompose at T > 300 °C, as indicated by the small weight loss observed in the TGA thermogram.
C present in the OA chain and usually detected at 284.4 eV.29 The results were inconsistent with the chemical composition of the surfactant (i.e. CC/COO = 5:1 instead of 2:1) and a significant worsening of the fitting quality was achieved. Therefore, we conclude that the component at 288.3 eV cannot be associated to remaining surfactant units but, more reasonably, derives from very small residual traces of unreacted zinc acetate precursor, which probably give rise to the weak bands observed in FT-IR spectra and decompose at T > 300 °C, as indicated by the small weight loss observed in the TGA thermogram.
These results support a fairly complete elimination of the organic surfactants after the washing treatment with EtOH.
Electrical measurements
In order to check the sensing properties of the obtained materials, ZnO particles were homogeneously mixed with EtOH until a dense mixture was obtained. Then, a few drops of this paste were deposited by drop-casting onto Suprasil quartz slides equipped with gold current collectors. As a representative example, Fig. 6a shows the SEM image of the deposited ER sub-microcrystals. It appears that the deposition does not modify the particle shape. Fig. 6b reports the resistance variation in films of ER ZnO crystals when pulses of the target gas (CO/air) with known CO concentration (62.5–500 ppm) were introduced into the measuring chamber, alternating with air pulses at 275 °C.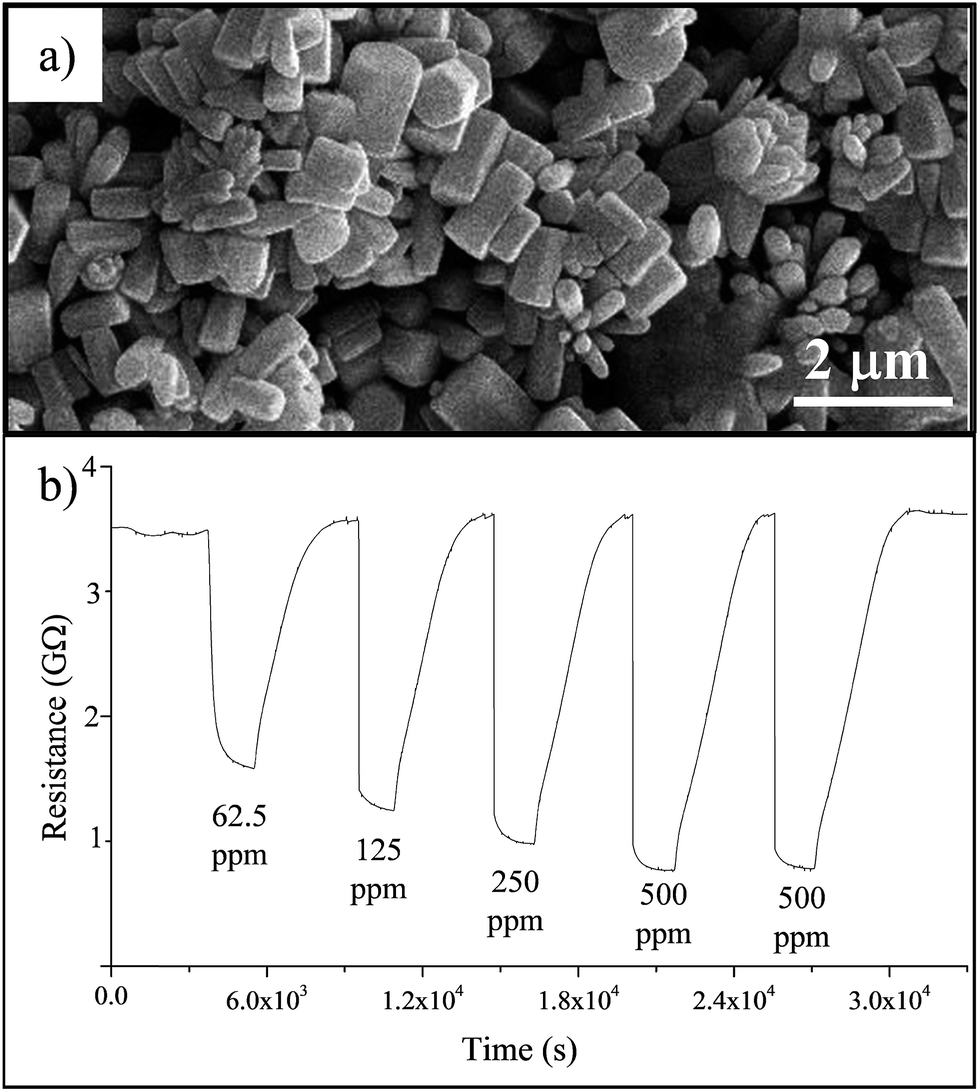 | ||
| Fig. 6 (a) SEM image of ER sub-microcrystals deposited by drop-casting onto a Suprasil quartz slide; (b) electrical resistance variation for ER films after exposure to different concentrations of CO (62.5–500 ppm) in dry air at 275 °C. | ||
Fig. 7a summarizes the electrical responses of shape controlled ZnO samples, under the highest CO concentration ([CO] = 500 ppm in dry air), as a function of the operating temperature. ER sub-microrods show better sensitivity at each operating temperature with the highest S values of 4.2 at 200 and 225 °C. Above these temperatures, their responses slowly decrease. EP and PY large microcrystals display instead very low electrical responses, which are basically unaffected by the change of the operating temperature. It must be reported that we did not perform sensing experiments above 350 °C, since particle coarsening and aggregation phenomena occur, leading to substantial changes in the morphology of the crystals.
 | ||
| Fig. 7 (a) Electrical responses of the morphology controlled ZnO crystals as a function of the operating temperature recorded under 500 ppm [CO] in dry air. (b) Electrical responses (S = RAIR/RMIX) of differently shaped ZnO crystals recorded under 500 ppm [CO] in dry air at 250 °C as a function of their total surface area (SSABET for ER and the geometric SSAGEO for PY and EP samples) and the area of their high-energy (0001) exposed crystal faces (c) sensing performances of ZnO® samples compared with those of shape controlled ZnO particles under the same operating conditions. | ||
Since it is well known that the sensing properties are affected by the surface area but also depend on the structure of the exposed crystal surfaces,1–3,5,6 the electrical responses of shape controlled ZnO crystals under CO (500 ppm)/air at 350 °C were plotted against the total surface area (SSABET for ER and the geometric SSAGEO for PY and EP samples) and the area of the highest-energy (0001) exposed crystal faces (Fig. 7b). It appeared that the performances of ZnO particles improve as either the total surface area or the area of their high energy facets increase. This indicates that the sensing properties of shape controlled ZnO crystals are attributable both to the total surface area and to the exposition of high-energy surfaces.
Hence, in order to better understand the role of the exposed crystal facets in upgrading the sensing properties, the electrical behaviors of the ZnO solvothermal samples have been compared with those of commercial zinc oxide (ZnO®) nanoparticles having uneven morphological features, but significantly larger SSABET (see Morphological characterization).
According to their higher porosity ZnO® nanocrystals displays better sensing performance than PY and EP microcrystals, with a maximum electrical response of 2.6 at 275 °C (Fig. 7c).
Unexpectedly, their sensing behavior at each operating temperature is worse than that observed for ER faceted sub-microcrystals, which have lower specific surface area, but display a relevant area of (0001) surfaces (see Table 1).
These results suggest that the presence of (0001) high-energy faces represents a fundamental requirement to upgrade the sensitivity of the ZnO layers16,17 and that the synergistic combination of appropriate crystal faces and high surface area should be an effective way to improve the ZnO sensitivity. On the same basis it is fully justified the low electrical response of EP and PY nanocrystals (Fig. 7a and c) which, do not or slightly expose the (0001) surfaces, respectively.
Finally, it must be observed that ER crystals display a relatively constant electrical response for a wide range of operating temperatures and, in comparison to ZnO® nanoparticles, they require a lower activating temperature for obtain reliable sensitivity toward CO (Fig. 7c).
ESR investigation
In previous studies,18–20 we suggested a sensing mechanism of ZnO®, based on the ESR detection of paramagnetic species associated to electrical resistance.Thus to investigate how the morphological features (i.e. size and shape) affect the formation and the reactivity of the paramagnetic species (typically singly ionized oxygen vacancies,  ; interstitial Zn,
; interstitial Zn,  ) involved in the ZnO sensing mechanism, a comprehensive ESR investigation was performed on shape controlled ZnO micro and sub-microcrystals (Fig. 8). The spectra were recorded under vacuum (p < 10−2 mbar) at 130 K, (i) after contacting samples with air, and (ii) under CO/Ar mixture (see Experimental section) at two temperatures (250 °C or 350 °C). We utilized CO/Ar mixture as gas probe instead of CO/air, in order to distinguish the electronic effects due to reducing gas from that due to the oxidizing one.
) involved in the ZnO sensing mechanism, a comprehensive ESR investigation was performed on shape controlled ZnO micro and sub-microcrystals (Fig. 8). The spectra were recorded under vacuum (p < 10−2 mbar) at 130 K, (i) after contacting samples with air, and (ii) under CO/Ar mixture (see Experimental section) at two temperatures (250 °C or 350 °C). We utilized CO/Ar mixture as gas probe instead of CO/air, in order to distinguish the electronic effects due to reducing gas from that due to the oxidizing one.
 | ||
| Fig. 8 Experimental ESR spectra of ZnO microcrystals at 130 K: (a) samples treated in air for 30 min at 250 °C; the insets show the corresponding shapes of the particles and the signal related to zinc vacancies; (b) after treatment under CO (500 ppm)/Ar mixture stream for 30 min at 250 °C; (c) after treatment under O2 at RT for 10 min and then vacuum (p < 10−2 mbar). The inset in (b) and (c) show the reactions occurring during CO/Ar and O2 treatments. | ||
As reported by Gurlo et al.,30 the paramagnetic defects above mentioned cannot be observed by ESR at the sensor operating temperatures. Hence, powders have been treated in situ (at the sensor operating temperature), and the spectra recorded at low temperature after evacuation and rapid quenching of the samples.
A weak low-field signal with g-factor close to the free-electron value (g = 2.0023) and a broad strong resonance line centered at g = 1.962 are easily detectable in all samples after treatment under air stream (80 cm3 min−1) at 250 °C (Fig. 8a). The shape and the intensity of the ESR signals are nearly the same in the as-prepared powders (spectra not shown). After subjecting the powders to CO/Ar atmosphere, the intensity of this latter signal remarkably decreases in ER and EP samples, while it remains almost unchanged for PY microcrystals (Fig. 8b).
Finally, oxygen contact at 298 K (Fig. 8c) induces a slight recovering of the spectral lines, and no other signals (e.g. O2−˙ species) have been observed in the spectrum.
In the ESR spectra acquired on samples subjected to initial air stream 350 °C (not reported), no significant variations of the signals trend and intensity have been observed.
In the literature contrasting attributions have been reported for these signals and they were differently assigned to Zn interstitial centers and to bulk or surface oxygen vacancies.20,31–36 In a very recent report,31 the signal at g = 2.002 has been assigned to two mutually close zinc vacancies ( ). The higher field signal at g ≈ 1.96, has been recently attributed to shallow donor centers, e.g. Zn interstitial in the crystal lattice of ZnO.33,34 Some other studies report that it originates from unpaired electrons trapped in oxygen vacancies.20,34–36
). The higher field signal at g ≈ 1.96, has been recently attributed to shallow donor centers, e.g. Zn interstitial in the crystal lattice of ZnO.33,34 Some other studies report that it originates from unpaired electrons trapped in oxygen vacancies.20,34–36
In order to attribute these signals as fairly as possible, we compared the ESR spectra of shape controlled ZnO with those of commercial ZnO nanoparticles (ZnO®), which have been already reported in our previous studies.18–20
On this purpose, we repeated the ESR investigation on ZnO® under the same conditions experienced by morphology controlled ZnO samples (Fig. 9).
 | ||
| Fig. 9 Experimental ESR spectra of ZnO® nanocrystals recorded under vacuum (p < 10−5 mbar) at 130 K after subjecting the samples to air, CO/Ar mixture and O2 at (a) 250 °C and (b) 350 °C. | ||
Either at RT or after treatment in air at 250 °C, two very close and weak resonance lines at g = 1.959 and g = 1.955 are detectable (Fig. 9a). The overall amount of defects in ZnO® (in the range of 1013 spin cm−1) is much lower than that observed in ER, EP and PY crystals (in the range of 1018 spin cm−1 for EP). Such signals are identical to those previously assigned to singly ionized oxygen vacancies  and to
and to  interstitial centres.18
interstitial centres.18
These paramagnetic species are generated by the reactions:
 | (4) |
 | (5) |
Under CO/Ar atmosphere at 250 °C (Fig. 9a), a significant growth of the  concentration has been observed, and attributed to the reactions:
concentration has been observed, and attributed to the reactions:
 | (6) |
 | (7) |
Conversely, the concentration of  does not significantly change.
does not significantly change.
The  resonance lines undergo a significant depletion after interaction with molecular oxygen at 298 K (Fig. 9a), possibly related to a partial re-oxidation of zinc interstitial species.
resonance lines undergo a significant depletion after interaction with molecular oxygen at 298 K (Fig. 9a), possibly related to a partial re-oxidation of zinc interstitial species.
At 350 °C after air (Fig. 9b) the spectra of ZnO® nanoparticles show the exclusive presence of the resonance lines attributed to singly ionized surface oxygen vacancies  . The concentration of these species sharply increases under CO/Ar atmosphere. This indicates that new oxygen vacancies have been created according to the following reactions:
. The concentration of these species sharply increases under CO/Ar atmosphere. This indicates that new oxygen vacancies have been created according to the following reactions:
 | (8) |
After O2 contact, the amount of  decreases and no other features (e.g. O2−˙ signal) have been observed in the spectrum.
decreases and no other features (e.g. O2−˙ signal) have been observed in the spectrum.
The results obtained by ESR spectra performed on ZnO® nanoparticles compared to those of shape controlled ZnO crystals highlight that the type, the amount and the reactivity of the paramagnetic defects are strongly related to the particle morphology. Therefore, also the sensing mechanism has to be discussed referring to their particular defectivity.
Along this direction, to better relate the sensing mechanism to the paramagnetic species, we also performed UV-irradiation experiments on ER, EP and PY crystals and commercial ZnO particles. This test was carried out in order to attribute the defects in shape controlled ZnO and, in particular, to verify if the signal at g = 1.962 (Fig. 8) was due to Zn interstitial atoms. In fact, Ischenko et al.32 observed that the concentration of Zn interstitial atoms increases upon UV-light exposure. Accordingly, the concentration of  interstitial centres in ZnO® raises under UV irradiation (Fig. S4, ESI†). Conversely, no significant changes of the spectral lines were observed for ER, EP and PY crystals.
interstitial centres in ZnO® raises under UV irradiation (Fig. S4, ESI†). Conversely, no significant changes of the spectral lines were observed for ER, EP and PY crystals.
Taking into account all the above outcomes, we may suggest that the signal at g = 1.962 in ER, EP and PY spectra is more likely related to electrons in singly ionized oxygen vacancy  .
.
In several previous studies on metal oxide gas sensors,5,7–9 we attributed great mechanistic relevance to oxygen defects, suggesting that the variation of electrical response occurring after interaction with the reducing gases would be originated by the alternate formation and filling of oxygen vacancies (vacancy mechanism) during the gas pulse. In particular, on the basis of a comprehensive ESR investigation, we evidenced that the  amount raises in connection with the reducing gas treatment, while it decreases in the presence of oxidation treatments. This mechanism was validated for SnO2 nanocrystals5–9 and also for ZnO® nanoparticles.18–20
amount raises in connection with the reducing gas treatment, while it decreases in the presence of oxidation treatments. This mechanism was validated for SnO2 nanocrystals5–9 and also for ZnO® nanoparticles.18–20
Unexpectedly, in the case of shape controlled ER and EP ZnO crystals, we observed an inverse reactivity of  species: their amount decreases under CO/Ar atmosphere while it progressively increases during the oxygen treatment (Fig. 8b and c). This suggests that the vacancy mechanism is not prevalent in ER and EP crystals.
species: their amount decreases under CO/Ar atmosphere while it progressively increases during the oxygen treatment (Fig. 8b and c). This suggests that the vacancy mechanism is not prevalent in ER and EP crystals.
These particles, beside exposing the {1010} non-polar planes with low surface energy, possess a relatively high surface area of O-terminated (000−1) (i.e. bottom) and the Zn-terminated (0001) (i.e. top) polar planes with high surface energy. The intrinsic dipole generated and the presence in (0001) of zinc atoms coordinatively unsaturated in three-fold and two-fold coordinated sites,16,17 lead to a higher chemisorption of oxygen anionic species (e.g. O−, O2−) on this surfaces. This on one hand preserves the surfaces from the formation of further oxygen defects, and on the other produces reactive oxygen species available for the reactions with the reducing gas. The subsequent oxygen-consuming processes generate electrons which raise the conductance of the oxide sensing layer, boosting the sensing properties of ER sub-microcrystals with highly exposed (0001) surfaces. Furthermore, the release of electrons may induce a back-shift of the VO ionization equilibrium (reaction (4)), in ER and EP crystals after CO/Ar contact, with consequent decrease of  species (Fig. 8b).
species (Fig. 8b).
In summary, monitoring paramagnetic defects helps to better describe the sensing action, highlighting the predominance of a specific sensing mechanism in shape controlled ZnO particles.
Conclusions
The present study comprehensively explores the sensing behavior toward CO of solvothermally obtained shape controlled ZnO, having specific exposed crystal surfaces, in comparison to that of commercial ZnO (ZnO®) with nanometric size and uneven shape.The performances have been discussed referring to the paramagnetic species ( and
and  ) involved in the sensing, in order to associate defects amount and reactivity to the particles morphology and, in turn, to a specific sensing mechanism.
) involved in the sensing, in order to associate defects amount and reactivity to the particles morphology and, in turn, to a specific sensing mechanism.
In particular, the ESR investigation revealed that:
• in ZnO®, upon interaction with CO, the amount of  species raises and successively decreases in the presence of oxidation treatments. This supports our previous hypothesis about a sensing action essentially related to the alternate formation and filling of surface oxygen vacancies during the gas pulse.
species raises and successively decreases in the presence of oxidation treatments. This supports our previous hypothesis about a sensing action essentially related to the alternate formation and filling of surface oxygen vacancies during the gas pulse.
• in the case of shape controlled ER and EP ZnO crystals, an inverse reactivity of oxygen paramagnetic defects have been detected: their amount decreases under CO/Ar atmosphere while it progressively increases during the oxygen treatment.
We explained these results by suggesting that the in ER and EP shape controlled crystals the ionosorption mechanism prevails, due to their peculiar exposition of (0001) crystal faces. These surfaces are terminated with zinc atoms coordinatively unsaturated, which promote a higher chemisorption of oxygen anionic species (e.g. O−, O2−). This on one hand may hinder the formation of  species at the surface of the oxide, and on the other supplies oxygen anions for the reactions with the reducing gas.
species at the surface of the oxide, and on the other supplies oxygen anions for the reactions with the reducing gas.
In summary, the above findings may positively contribute to the debate still existing between the oxygen vacancy and ionosorption models by suggesting a better localization of the sensing action and the occurrence of morphology-dependent sensing mechanisms in ZnO.
Acknowledgements
This work has been performed in the frame of the European COST action MP1202 “Rational design of hybrid organic inorganic interfaces: the next step towards advanced functional materials”. The authors personally thank Michele Guerini for his support in the experimental work, Prof. Riccardo Ruffo for valuable discussions concerning on electrical measurements and Dr Paolo Gentile for his assistance with SEM investigation, Dr Lidia Armelao and Dr Marzio Rancan for XPS analysis. MR thanks Corimav for its support within the PCAM European Doctoral Programme.References
- A. Gurlo, Nanoscale, 2011, 3, 154–165 RSC.
- X. Wang, X. Han, S. Xie, Q. Kuang, Y. Jiang, S. Zhang, X. Mu, G. Chen, Z. Xie and L. Zheng, Chem.–Eur. J., 2012, 18, 2283–2289 CrossRef CAS PubMed.
- Q. Kuang, X. Wang, Z. Jiang, Z. Xie and L. Zheng, Acc. Chem. Res., 2014, 47, 308–318 CrossRef CAS PubMed.
- M. R. Alenezi, A. S. Alshammari, K. D. G. I. Jayawardena, M. J. Beliatis, S. J. Henley and S. R. P. Silva, J. Phys. Chem. C, 2013, 117, 17850–17858 CAS.
- M. D'Arienzo, D. Cristofori, R. Scotti and F. Morazzoni, Chem. Mater., 2013, 25, 3675–3686 CrossRef.
- M. D'Arienzo, L. Armelao, C. M. Mari, S. Polizzi, R. Ruffo, R. Scotti and F. Morazzoni, RSC Adv., 2014, 4, 11012–11022 RSC.
- M. D'Arienzo, L. Armelao, A. Cacciamani, C. M. Mari, S. Polizzi, R. Ruffo, R. Scotti, A. Testino, L. Wahba and F. Morazzoni, Chem. Mater., 2010, 22, 4083–4089 CrossRef.
- M. Acciarri, C. Canevali, C. M. Mari, M. Mattoni, R. Ruffo, R. Scotti, F. Morazzoni, D. Barreca, L. Armelao, E. Tondello, E. Bontempi and L. E. Depero, Chem. Mater., 2003, 15, 2646–2650 CrossRef CAS.
- L. Armelao, D. Barreca, E. Bontempi, F. Morazzoni, C. Canevali, L. E. Depero, C. M. Mari, R. Ruffo, R. Scotti and E. Tondello, Appl. Magn. Reson., 2002, 22, 89–100 CrossRef CAS.
- N. Yamazoe and N. Miura, in Chemical Sensor Technology, ed. S. Yamauchi, Elsevier, N.Y., 1992, vol. 4, p. 19 Search PubMed.
- C. Xu, J. Tamaki, N. Miura and N. Yamazoe, Sens. Actuators, B, 1991, 3, 147–155 CrossRef CAS.
- J. N. Huang, K. Matsunaga, N. Shimanoe, T. Yamazoe and T. Kunitake, Chem. Mater., 2005, 17, 3513–3518 CrossRef CAS.
- D. Degler, S. Wicker, U. Weimar and N. Barsan, J. Phys. Chem. C, 2015, 119, 11792–11799 CAS.
- A. Gurlo, ChemPhysChem, 2006, 7, 2041–2052 CrossRef CAS PubMed.
- Z. Wang, J. Xue, D. Han and F. Gu, ACS Appl. Mater. Interfaces, 2015, 7, 308–317 CAS.
- X. G. Han, H. Z. He, Q. Kuang, X. Zhou, X. H. Zhang, T. Xu, Z. X. Xie and L. S. Zheng, J. Phys. Chem. C, 2009, 113, 584–589 CAS.
- S. Tian, F. Yang, D. Zeng and C. Xie, J. Phys. Chem. C, 2012, 116, 10586–10591 CAS.
- F. Morazzoni, R. Scotti and S. Volontè, J. Chem. Soc., Faraday Trans., 1990, 86, 1587–1591 RSC.
- F. Morazzoni, R. Scotti and N. Minnaja, J. Chem. Soc., Faraday Trans., 1991, 87, 493–496 RSC.
- F. Morazzoni, R. Scotti, P. Di Nola, C. Milani and D. Narducci, J. Chem. Soc., Faraday Trans., 1992, 88, 1691–1694 RSC.
- C. T. Dinh, T. D. Nguyen, F. Kleitz and T. O. Do, ACS Nano, 2009, 3, 3737–3743 CrossRef CAS PubMed , and references therein.
- D. Briggs and M. Seah, Practical Surface Analysis, Wiley, Chichester, 1990 Search PubMed.
- D. A. Shirley, Phys. Rev. B: Condens. Matter, 1972, 5, 4709–4714 CrossRef.
- S. Brunauer, P. H. Emmet and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barret, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef.
- B. Ludi and M. Niederberger, Dalton Trans., 2013, 42, 12554–12568 RSC , and references therein.
- G. Munoz Hernandez, A. Escobedo-Morales and P. Umpada, Cryst. Growth Des., 2009, 9, 297–300 CAS.
- D. A. Schwartz, N. S. Norberg, Q. P. Nguyen, J. M. Parker and D. R. Gamelin, J. Am. Chem. Soc., 2003, 125, 13205–13218 CrossRef CAS PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, ed. G. Chastain, Perkin Elmer Corporation, Eden Prairie, Minnesota, 1992 Search PubMed.
- A. Gurlo and R. Riedel, Angew. Chem., Int. Ed., 2007, 46, 3826–3848 CrossRef CAS PubMed.
- Z. Bian, T. Tachikawa, P. Zhang, M. Fujitsuka and T. Majima, Nat. Commun., 2014, 5, 3038, DOI:10.1038/ncomms4038.
- V. Ischenko, S. Polarz, D. Grote, V. Stavarache, K. Fink and M. Driess, Adv. Funct. Mater., 2005, 15, 1945–1954 CrossRef CAS.
- H. Zeng, G. Duan, Y. Li, S. Yang, X. Xu and W. Cai, Adv. Funct. Mater., 2010, 20, 561–572 CrossRef CAS.
- X. Xue, T. Wang, X. Jiang, J. Jiang, J. Pana and Y. Wu, CrystEngComm, 2014, 16, 1207–1216 RSC.
- D. Liu, Y. Lv, M. Zhang, Y. Liu, Y. Zhu, R. Zonga and Y. Zhu, J. Mater. Chem. A, 2014, 2, 15377–15388 CAS.
- J. J. Schneider, R. C. Hoffmann, J. Engstler, S. Dilfer, A. Klyszcz, E. Erdem, P. Jakes and R. A. Eichel, J. Mater. Chem., 2009, 19, 1449–1457 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the morphological and spectroscopic features of solvothermal ZnO crystals are reported in this section. The ESR spectrum of ZnO® after UV-Vis irradiation is also included. See DOI: 10.1039/c6ra09824j |
| This journal is © The Royal Society of Chemistry 2016 |