DOI:
10.1039/C6RA09819C
(Paper)
RSC Adv., 2016,
6, 66468-66476
Selective electrochemical detection of dopamine in the presence of uric acid and ascorbic acid based on a composite film modified electrode†
Received
15th April 2016
, Accepted 1st July 2016
First published on 6th July 2016
Abstract
An electrochemical sensor was built by the layer-by-layer self-assembly method for selective and sensitive determination of dopamine (DA) in the presence of uric acid (UA) and ascorbic acid (AA), based on vanadium-substituted polyoxometalates, copper oxide and chitosan–palladium. This sensor was characterized by UV-vis spectroscopy, transmission electron microscopy (TEM), atomic force microscopy (AFM), scanning electron microscopy (SEM) and X-ray photoelectron spectroscopy (XPS). Cyclic voltammetry, electrochemical impedance spectroscopy and current–time were employed to investigate the electrochemical and sensing properties. The results indicated the proposed sensor had good electrocatalytic activity towards the oxidation of dopamine and exhibited a low detection limit of 0.045 μM (S/N = 3), a wide linear range of 0.25–217 μM and fast response (<2 s). In addition, the sensor exhibited excellent reproducibility, long-term stability and good anti-interference ability towards common coexistent compounds. Finally, this sensor was employed to detect DA in real blood serum samples and the recovery is acceptable within the allowed error range.
1. Introduction
Dopamine (DA) is one of important catecholamine neurotransmitters in the central nervous system of mammals. Malfunctions in the dopaminergic system may lead to serious diseases such as schizophrenia and Parkinson's disease.1,2 In addition, DA is available for intravenous medication, which acts on the sympathetic nervous system, to produce effects such as increasing heart rate and blood pressure.3,4 Therefore, monitoring the concentration of DA is vitally important both in vitro and in vivo. Many methods have been used to detect DA, for instance, chemiluminescence,5,6 ultraviolet-visible spectrometry,7,8 fluorimetry,9,10 capillary electrophoresis,11,12 liquid chromatography13,14 etc. Compared with these methods, the direct electrochemical method for DA detection has received considerable interest due to its simplicity, rapid response and low cost.15 However, direct redox reaction of DA at bare electrodes is irreversible and requires high overpotentials. And electrochemical determination of DA is also plagued by interference from the coexisting compounds such as uric acid (UA) or ascorbic acid (AA) in biological samples, because their oxidation potentials are close to that of DA on unmodified electrodes,1,3,16 which results in rather poor selectivity. Normally, the electrochemical behavior and electron transfer property of electrodes could be improved by introduction of active components. Various electrode-modified were developed by introduction of active materials to achieve high electrochemical activity, anti-interference, and lower overpotential, such as carbon nanotubes,16,17 graphene,18–20 gold nanoparticle,3,21 polymer22,23 and so on.
Polyoxometalates (POMs), as a well-known class of anionic metal-oxide clusters, are widely used as electrocatalysts and electrochemical sensors owing to their excellent chemical stability, reversible redox activities, and special electrocatalytic properties.24–30 Vanadium-substituted POMs as a member of POMs family are stable in a large pH domain including the physiological pH domain and the solutions of their oxidized (VV) and reduced (VIV) forms are stable in the open air.31,32 Hence, vanadium-substituted POMs are promising candidates for building electrochemical sensors. However, the low specific surface area of POMs is a drawback for electrocatalytic activity, thus good support materials for immobilizing POMs becomes an important issue.33,34
Nanomaterials are widely used because they have novel chemical and physical properties and high specific surface area that can provide more active sites. Copper oxide (CuO) is a p-type metal oxide semiconductor with a narrow band-gap that has been effectively used in the fabrication of electrical, optical and photovoltaic devices, heterogeneous catalysis, magnetic storage media and lithiumion electrode materials15,35 due to its virtue of natural abundance, low production cost, good electrochemical and catalytic properties. In particular, recent progress in CuO catalysis has revealed that CuO in nanostructures can enhance electrochemical response for DA.15,16,35 Pd nanoparticles are one of the most popular noble metal nanoparticles with higher abundance and lower cost in comparison with Pt and Au nanoparticles,36–39 and many characteristics including high surface to-volume ratio, good biocompatibility, conductivity and catalytic property.39–41 It is well known that ultrafine noble metal nanoparticles have improved catalytic properties due to their increased surface area and the number of edge and corner atoms.42,43 However, the ultrafine nanoparticles usually tend to aggregate in order to minimize the total surface energy.44,45 To avoid the aggregation, surfactants, polymers, dendrimers as well as different types of ligands can be used as capping agent to stabilize the loaded nanoparticles to achieve homogeneous dispersion. Chitosan is one of the most promising natural polymers for using as an immobilization matrix due to its excellent properties such as biodegradability, nontoxicity, biocompatibility, high mechanical strength, good film-forming properties, and low cost.46,47 Chitosan–palladium (Cs–Pd) has been studied extensively in catalytic fields,48–50 and there are just few reports about determination of DA.
In this work, a vanadium-substituted polyoxometalate, CuO and Pd nanoparticles were chosen as active electrode-modified materials to built an electrochemical sensor by the layer-by-layer self-assembly method for determination of DA. In comparison to the electrodes containing only one of these three active materials, a remarkable enhancement of voltammetric response of DA at lower applied potential +0.73 V was observed at this composite sensor, and meanwhile almost no voltammetric response of AA and UA was detected. The high selectivity and sensitivity of this proposed sensor are attributed to combined functions of these three active components. This proposed sensor was successfully applied for selective determination of DA in the presence of potential interference species of AA and UA in real blood samples.
2. Experimental
2.1. Reagents and chemicals
Copper acetate dihydrate [Cu(CH3COO)2·H2O], sodium hydroxide (NaOH, 96% purity), K2PdCl4 and chitosan (CS MW 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–300000) were purchased from Aladdin, poly(ethylenimine) (PEI MW 750000) was obtained from Aldrich. α2-K7P2VW17O62·18H2O (P2W17V) was prepared according to the literature.51 CuO, CS–Pd nanoparticles were prepared according to the literature52,53 and characterized by TEM (Fig. S1 and S2†). All the chemicals were analytical grade and used without further purification. The water used in all experiments was deionized to a resistivity of 18 MΩ cm.
000–300000) were purchased from Aladdin, poly(ethylenimine) (PEI MW 750000) was obtained from Aldrich. α2-K7P2VW17O62·18H2O (P2W17V) was prepared according to the literature.51 CuO, CS–Pd nanoparticles were prepared according to the literature52,53 and characterized by TEM (Fig. S1 and S2†). All the chemicals were analytical grade and used without further purification. The water used in all experiments was deionized to a resistivity of 18 MΩ cm.
2.2. Syntheses of CuO nanoparticles
In a typical synthesis process, 100 mL of 0.02 M copper acetate solution was mixed with 0.50 mL acetic acid and then slowly heated in a three-necked refluxing pot under stirring. When the temperature reached at 90 °C, 0.50 g NaOH was added, which resulted in black color solution (pH = 5–6) indicating the formation of CuO NPs. In this reaction condition, the NaOH acted as a basic source and the acetic acid breaks the precipitates for the formation of uniform and disperse CuO NPs. After 10–15 min refluxing, black-colored colloidal solution was obtained, followed by centrifugation at 3000 rpm for 2 min and washing with deionized water and ethanol.52
2.3. Syntheses of CS–Pd nanoparticles
In a typical procedure, a K2PdCl4 (50 μL, 20 mM) aqueous solution was mixed with chitosan (3 mL, 2.0 mg mL−1), the mixtures were stirred for 30 min, then freshly prepared aqueous solutions of NaBH4 (50 μL, 0.2 M) were added quickly to the mixture, and kept stirring for another 90 min until the entire reduction of metal salts. The resulted nanocomposites were kept at room temperature for characterization.53
2.4. Apparatus
UV-vis absorption spectra were recorded on a quartz slide using a U-3900 UV-vis spectrophotometer from Hitachi (Japan) and used to detect the growth of the composite film. The element compositions of the composite film were identified by X-ray photoelectron spectra (XPS), which were performed on an ESCALAB-MKII spectrometer with Mg Kα X-ray radiation as the X-ray source for excitation. Surface morphology and topography were examined by scanning electron microscopy (SEM) and atomic force microscopy (AFM). SEM images were obtained with S-4300, AFM was taken by Dimension TM3100 series of AFM produced from Digital Instruments (Santa Barbara, CA, USA). Transmission electron microscope (TEM) image was obtained on an H-7650 TEM. All the electrochemical experiments were performed on a CHI760D electrochemical workstation with the ITO electrode coated by the self-assembled film as the working electrode, Ag/AgCl (3 M KCl) as the reference electrode and platinum coil as the counter electrode.
2.5. Fabrication of the composite film
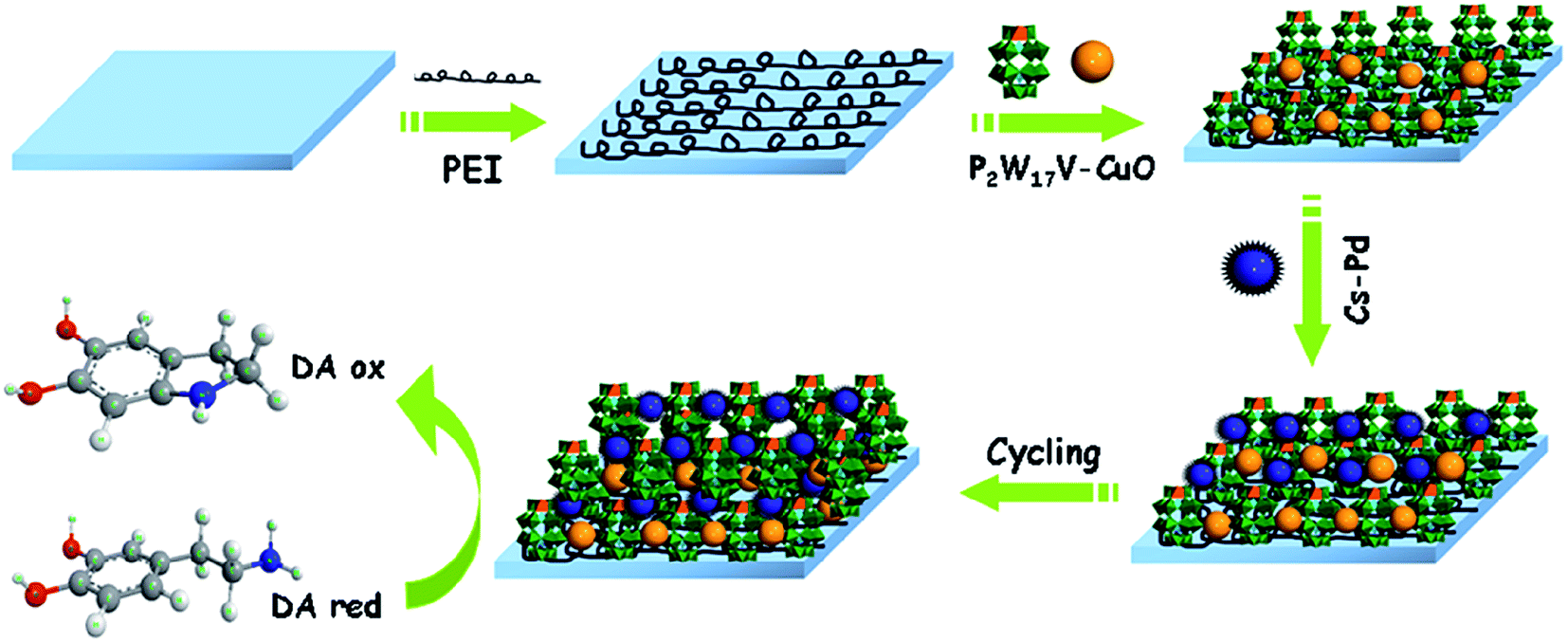
The substrates (silica, quartz glass slide or ITO) were treated according to the literature.54 The silicon wafers were cleaned respectively in a 40 °C propanol, ethanol absolute and deionized water bath for 20 min. The quartz slides were cleaned in Piranha solution (H2SO4:H2O2 = 7:3 v/v) for 20 min at 80 °C, and rinsed with deionized water. Further purification was carried out by immersing the slides in an H2O/H2O2/NH3OH (5:1:1) (v/v/v) bath for 20 min at 70 °C, thoroughly rinsed with deionized water, and dried under a nitrogen stream. The ITO glass slides were continuously immersion in a series of ultrasonically agitated solvents (30% KOH alcohol solution, ethanol, H2O) at 40 °C for 20 min. Then they were rinsed with deionized water and dried under a nitrogen stream. The clean substrates were immersed in 10 mM PEI solution overnight before using, followed by washing with deionized water, dried under a nitrogen stream. Then the PEI-coated substrates were alternately immersed in 2 mM P2W17V–CuO and CS–Pd for 20 min. After each immersion, the substrates were rinsed with water and dried with nitrogen gas. Repeating the recycle, the composite film {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} was successfully fabricated on ITO. Scheme 1 illustrates the fabrication process of the proposed sensor.
 |
| | Scheme 1 The schematic illustration of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} proposed sensor. | |
3. Results and discussion
3.1. UV-vis absorption spectra
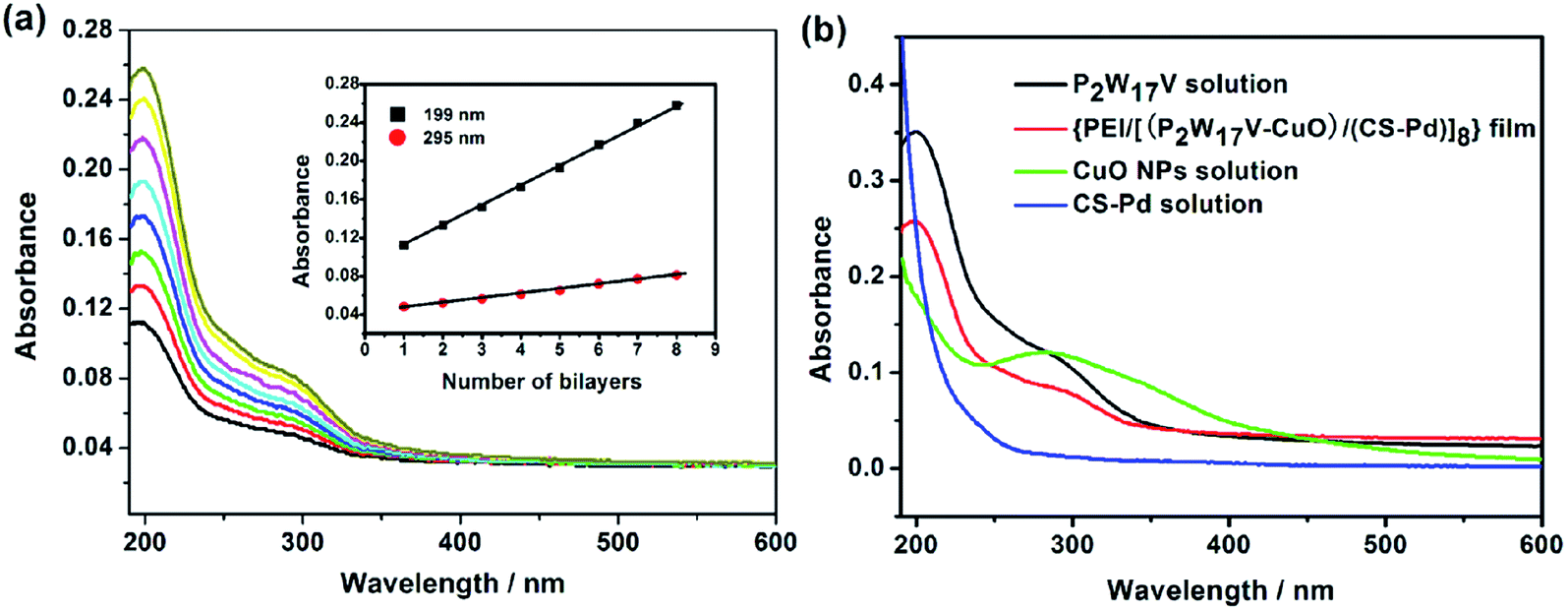
UV-vis spectroscopy has proved to be a useful and facile technique for evaluating the growth process of multilayers, and thus used in the present work to monitor the LBL assembly process of {PEI/[(P2W17V–CuO)/(CS–Pd)]n} multilayer film. Fig. 1a shows the UV-vis spectra of {PEI/[(P2W17V–CuO)/(CS–Pd)]n} films (with n = 1–8) assembled on a quartz slide. The spectra show two characteristic absorption peaks at 199 and 295 nm, which are owing to the terminal oxygen to tungsten (Od → W) charge transfer transition and an overlap of CuO and P2W17V bands (see Fig. 1b), respectively. However, there is a slight red shift of about 3 nm for the band at 295 nm compared to that of the P2W17V solution (at 292 nm) (see Fig. 1b). This may be related to the broad band corresponding to CuO in the wavelength range of 230–420 nm. Furthermore, it can be seen from the spectrum there is no plasmon absorbance could be observed for CS–Pd solution between 190 and 600 nm. The inset of Fig. 1a displays the absorbance values at 199 nm and 295 nm increased linearly with the number of bilayer, indicating that the growth of the films for each deposition cycle was reproducible and the multilayer films had been constructed uniformly and homogeneously.
 |
| | Fig. 1 (a) UV-vis spectra of the {PEI/[(P2W17V–CuO)/(CS–Pd)]n} film with n = 1–8 (from bottom to top) for the quartz substrate (on both sides). The inset: plots of the absorbance values at 199 and 295 nm against bilayer number n; (b) UV-vis spectra of several solutions and the composite film. | |
3.2. X-ray photoelectron spectroscopy
The chemical composition of {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} multilayer film was analyzed by XPS (Fig. S3†). The survey spectrum shows the presence of W, V, Cu, Pd, C and N elements in the film. The peaks of W 4f7/2/W 4f5/2 at 34.97/37.12 eV and the peaks of V 2p3/2/V 2p1/2 at 515.92/522.85 eV are attributed to P2W17V (Fig. S3a and b†).55 The Cu 2p core level presents the doublet bonds at a binding energy of 934.08 and 953.78 eV (Fig. S3c†), corresponding to 2p3/2 and 2p1/2 of Cu element, which is ascribe to CuO.56 Additionally, the peaks of Pd and C are detected at 335.36, 338.27 and 284.09 eV, respectively (Fig. S3d and e†). They are assigned to palladium-chitosan nanocomposites.49,50,57 Spectrum of N 1s bond at 399.08 eV results from PEI (Fig. S3f†).54,58 XPS results further confirmed that PEI, P2W17V, CuO and Cs–Pd have been incorporated into the multilayer film.
3.3. Investigation of surface morphology
The topography and surface morphology of the {PEI/[(P2W17V–CuO)/(CS–Pd)]n/(P2W17V–CuO)} multilayer film were examined by AFM and SEM. Fig. 2 shows the AFM image of the {PEI/[(P2W17V–CuO)/(CS–Pd)]3/(P2W17V–CuO)} film deposited on silicon. The surface of the composite film consists of a large number protuberant peaks with a root-mean-square roughness of 2.34 nm calculated over an area of 1 μm × 1 μm.
 |
| | Fig. 2 AFM image of the {PEI/[(P2W17V–CuO)/(CS–Pd)]3/(P2W17V–CuO)}composite film. | |
SEM was performed to further reveal the morphology. As shown in Fig. 3a, isolated snowflake-like structure was observed on the surface of the film, which could be attributed to Pd-chitosan.53 In addition, the cross-section morphology of the multilayer film was observed in Fig. 3b with the thickness of about 200 nm.
 |
| | Fig. 3 The surface (a) and cross-section (b) SEM images of the composite film {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} on an ITO-coated glass. | |
3.4. Electrochemical behavior of the composite film
To study the electrochemical behavior of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film, cyclic voltammograms was first investigated in 0.2 M PBS buffer solution (pH = 7.0). As shown in Fig. 4a, three pairs of well-defined redox peaks (I/I′, II/II′ and III/III′) were observed in the potential region of −0.9 to 0 V vs. Ag/AgCl. The average of the oxidation and reduction peak potentials are −0.420 V (I/I′), −0.535 V (II/II′) and −0.770 V (III/III′), respectively. Redox peaks I and II are corresponding to two one-electron tungsten-centered (WVI → WV), and redox peak III is corresponding to two-electron tungsten-centered (WVI → WV).59 The inset of Fig. 4a shows the cyclic voltammogram of the composite film at positive potential range attributed to the redox peak of V center, which is corresponding to one one-electron redox process (VV → VIV).59–61 When the scan rate increases from 100 to 300 mV s−1, the potentials of cathodic peak (Epc) shift to negative potential values while the potentials of anodic peak (Epa) shift to positive potential values, which is consistent with a reversible but non-ideal redox process. Taking the peak II as a representative, the peak currents are linear with the scan rate as shown in Fig. 4b, indicating that electron transfer of the surface-confined redox couples is not diffusion limited.62,63 The surface coverage of P2W17V anions can be calculated according to the following formula:where ip is the peak current (amperes), n is the number of electrons transferred per electroactive species, ν is the scan rate (volts per second), A is the geometric area of the electrode (square centimeters), and all other terms have their usual significance.62,64 Taking the second couple of redox peaks for example, the surface coverage of P2W17V per layer amounts to 1.18 × 10−10 mol cm−2 for the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film by analyzing cyclic voltammograms at scan rate of 0.3 V s−1.
 |
| | Fig. 4 (a) Cyclic voltammograms of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film at negative potential at different scan rates from 100 to 300 mV s−1. The inset: the cyclic voltammogram of the composite film at positive potential range (0.2 M PBS (pH = 7.0) vs. Ag/AgCl); (b) plots of the cathodic and anodic peak currents of the couple waves II against scan rate. | |
Electrochemical impedance spectroscopy (EIS) characterization was performed to explore regarding the facile electron transfer behavior. The experiments were carried out with ITO/{[PEI/P2W17V]7}, ITO/{[PEI/(P2W17V–CuO)]7} and ITO/{PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} in a frequency range 0.001–100000 Hz with signal amplitude of 5 mV in 0.2 M PBS (pH 7.0). It is well known that the semicircle portion observed at high frequencies is attributed to the charge transfer limiting process in electrochemical impedance spectroscopy, which indicates the ability of electron transfer of different electrodes.65 As can be seen in Fig. 5, the diameter of the semicircle increases in the order of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} < {[PEI/(P2W17V–CuO)]7} < {[PEI/P2W17V]7} films, which demonstrates that the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film exhibits lower electron transfer resistance and greater electron transfer rate. This result also shows that CuO and CS–Pd improve the conductivity and the electron transfer process. The inset of Fig. 5 displays the equivalent circuit including the following: Rs is the ohmic resistance of the electrolyte solution; R1 is the resistance to charge transfer. It was assumed that the resistance to charge transfer (R1) was in parallel to the constant phase element (CPE), which was selected to replace the pure capacitor in the equivalent circuit by considering the surface roughness and inhomogeneity of the multilayer film. This parallel combination of R1 and CPE gave rise to a semicircle in the complex plane plot of Zim against Zre. According to the impedance spectrum, R1 was 3050 Ω and CPE was 4.38 × 10−4 F. The electric charge transmission speed constant (k) can be calculated by the following formula:
where
R1 is the resistance to charge transfer,
n is the number of electrons transferred per electroactive species,
C is the constant phase element (CPE),
A is the geometric area of the electrode (square meters), and all other terms have their usual meaning.
66,67 According to the formula
(2), the electron charge transfer rate constant (
k) of the {PEI/[(P
2W
17V–CuO)/(CS–Pd)]
7/(P
2W
17V–CuO)} composite film was estimated to be 1.28 × 10
−3 cm s
−1.
 |
| | Fig. 5 The EIS curves of the films {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)}, {[PEI/(P2W17V–CuO)]7} and {[PEI/P2W17V]7} in 0.2 M PBS (pH 7.0). The inset: a simple equivalent circuit about the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} electrode system. | |
3.5. The electrocatalytic property of the composite film
It is well known that dopamine exists in the physiological systems in the pH range of 6.0–8.0, so the influence of solution pH value on the oxidation of DA was investigated in 0.2 M KH2PO4 + Na2HPO4 solution from pH 6.0 to 8.0, which was assessed according to catalytic efficiency. The catalytic efficiency was calculated as defined by the equation:| | |
CAT = 100% × [Ip(POM, substrate) − Ip(POM)]/Ip(POM)
| (3) |
where Ip(POM, substrate) and (POM) are the catalytic currents of the POM in the presence and absence of substrate DA, respectively.68,69 As shown in Fig. 6, the catalytic efficiencies of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} modified electrode reached the maximum at pH 7.0, and hence pH 7.0 phosphate buffer solution was used for further studies.
 |
| | Fig. 6 Catalytic efficiency for the composite film {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} to 0.05 mM DA in different pH buffer solutions (pH = 6, 6.5, 7, 7.5 and 8). Scan rate: 50 mV s−1. | |
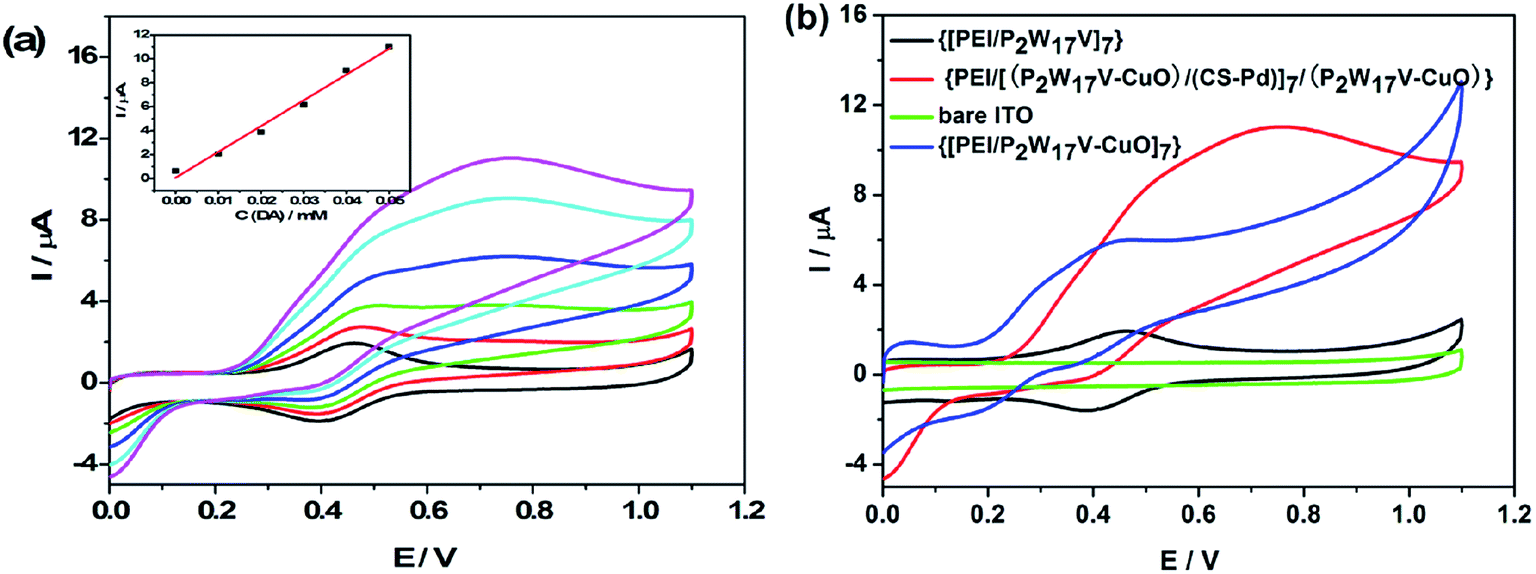
Fig. 7a shows cyclic voltammograms of {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} modified electrode in the presence of different concentration of DA in 0.2 M PBS (pH = 7.0) at a scan rate of 50 mV s−1. It can be seen that the oxidation peak current increases with the increase of DA concentration form 0.01 mM to 0.05 mM, which reveals a catalyze oxidation reaction of DA, the inset of Fig. 7a shows the dependence curves of oxidation peak currents on concentration of DA. The peak currents increase proportionally with DA, which demonstrates stable electrocatalytic activity and can be utilized for constructing a dopamine sensor. Fig. 7b shows cyclic voltammetry curves of bare ITO, {[PEI/P2W17V]7}, {[PEI/(P2W17V–CuO)]7} and {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} in 0.2 M PBS (pH = 7.0) containing 0.05 mM DA at a scan rate of 50 mV s−1. It can be seen that the current of {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film is much larger than the others, which indicates that the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film shows high catalytic activity towards dopamine. That may be attributed to the synergistic effects among three active components. Firstly, CuO and Cs–Pd nanoparticles with high surface-to-volume ratio could increase immobilized amount of P2W17V with inherent outstanding redox activity on the surface of the modified electrode to provide more active sites for DA molecules to adsorb and react. Secondly, highly conductive CuO and Pd nanoparticles could decrease the charge transfer resistance at the surface of electrode and accelerate electron transfer rate (see Fig. 5), which facilitated the electron transfer between DA and the electrode surface.
 |
| | Fig. 7 (a) Cyclic voltammograms of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} film with different concentrations of DA (0.01, 0.02, 0.03, 0.04 and 0.05 mM); (b) compared cyclic voltammograms of {[PEI/P2W17V]7} (black line), {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film (red line), {[PEI/P2W17V–CuO]7} (blue line) and bare ITO (green line) with 0.05 mM of DA in 0.2 M PBS (pH = 7.0). Scan rate: 50 mV s−1. | |
3.6. Sensing performance of the proposed sensor
3.6.1. The selectivity and sensitivity of the proposed sensor. Selectivity and sensitivity are important factors for sensing applications and usually affected by the applied potential. The amperometric responses of some possible interference species such as AA, UA, L-cysteine, H2O2 and glucose along with DA were measured by the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} proposed sensor in the 0.2 M PBS (pH 7.0) at different applied potentials as shown in Fig. 8. In the range of 0.4–0.7 V, the more positive the potential was applied, the higher sensitivity of the sensor for detection of DA showed, and at applied potential of 0.73 V, the sensitivity is maximum. On the other hand, in the applied potential range of 0.8–0.9 V, the proposed DA sensor was free from the interference of AA, but obvious current response of UA was observed; in the applied potential range of 0.4–0.6 V, the proposed DA sensor was free from the interference of UA, but the current response of AA was observed. At applied potential of 0.73 V, AA, UA, and other tested interfering species did not show observable response to DA, namely selectivity of the proposed sensor is highest. At 0.73 V, both selectivity and sensitivity were maximal, so 0.73 V was selected as the optimized applied potential. Moreover, we further analyzed some reductive compounds in serum and some inorganic ions such as Na+, K+, Ca2+, NO2− at applied potential of 0.73 V as shown in Fig. S4.† The result indicated the composite film modified film exhibited good anti-interference ability to these interference species.
 |
| | Fig. 8 The selectivity profile of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film obtained with DA (0.01 mM), AA (1 mM), UA (1 mM), L-cysteine (1 mM), H2O2 (1 mM) and glucose (1 mM) at different applied potentials in 0.2 M PBS (pH 7.0). | |
3.6.2. Amperometric detection of DA. The amperometric response of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film to the addition of DA was carried out in 0.2 M PBS (pH = 7.0) at an applied potential of +0.73 V. As seen from the inset of Fig. 9a, the current signal enhanced step-wise with each addition of DA, then reached a steady-state current (95% of the maximum) in less than 2 s, indicating a fast amperometric response behavior. The current responses were linear with DA concentrations from 0.25 to 217 μM (Fig. 9b), with the regression equation as: IDA (μA) = 1.81516 + 0.91874 × CDA (μM) (R2 = 0.99861). The sensitivity of the composite film for detecting DA was calculated to be 0.92 μA μM−1 and the detection limit was 0.045 μM at a signal-to-noise ratio of 3.
 |
| | Fig. 9 (a) Typical amperometric responses of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film to successive additions of several concentrations of DA at applied potential of +0.73 V vs. Ag/AgCl in 0.2 M PBS (pH 7.0); (b) calibration plot of steady-state currents obtained at the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film against concentrations of DA. | |
The sensing performances of the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film modified ITO electrode were more superior then most DA sensors which were listed in Table 1.
Table 1 A comparison of the sensing characteristics of the proposed sensor with some other sensors for determination of dopamine
| Electrode |
Linear range (M) |
LOD (μM) |
Ref. |
| GP-MWCNTs-AuNCs/GCE |
2 × 10−6 to 2.13 × 10−4 |
0.67 |
17 |
| Graphene/chitosan/GCE |
1 × 10−7 to 1.4 × 10−4 |
0.05 |
18 |
| PImox-GO/GCE |
1.2 × 10−5 to 2.78 × 10−4 |
0.63 |
20 |
| Methoxypolyethylene glycols/GCE |
2 × 10−6 to 1.4 × 10−4 |
0.0468 |
22 |
| p-Sulphonatocalix[6]arene/polypyrrole/platinum |
7.5 × 10−5 to 1.0 × 10−3 |
20 |
23 |
| PMo11V@GFs/GCE |
2 × 10−6 to 3 × 10−4 |
0.88 |
33 |
| Binuclear copper(II) complex/Au |
2 × 10−7 to 3 × 10−5 |
0.08 |
70 |
| GO/GCE |
1 × 10−6 to 1.5 × 10−5 |
0.27 |
71 |
| RGO/Pd-NPs/GCE |
1 × 10−6 to 1.5 × 10−4 |
0.233 |
72 |
| Rod shaped CuO/CPE |
3 × 10−7 to 1.4 × 10−6 |
0.18 |
73 |
| Poly-Evans Blue/GCE |
1 × 10−6 to 3 × 10−5 |
0.25 |
74 |
| FGGE |
5 × 10−7 to 5 × 10−5 |
0.25 |
75 |
| N-RGO/MnO/GCE |
1 × 10−5 to 1.8 × 10−4 |
3 |
76 |
| PoPD/E-RGO/GCE |
1 × 10−5 to 4 × 10−4 |
7.5 |
77 |
| 4 × 10−4 to 8 × 10−4 |
| MWCNTs/Nafion/ITO |
1 × 10−7 to 1 × 10−5 |
0.2 |
78 |
| Dopamine grafted ERG/PMB/GCE |
9.6 × 10−7 to 7.68 × 10−6 |
1.03 |
79 |
| {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)}/ITO |
2.5 × 10−7 to 2.17 × 10−4 |
0.045 |
This work |
3.6.3. Reproducibility and stability of the proposed sensor. The reproducibility was investigated from the current response to 0.05 mM DA with ten electrodes that were prepared under the identical conditions, and the relative standard deviations (RSD) were 4.26%, suggesting excellent reproducibility of the sensor prepared. In addition, redox reactions on the surface of sensor often cause sensor degradations, leading to a loss of activity. So the stability of the sensor was evaluated by cyclic voltammetry scanning for 100 cycles in 0.2 M PBS solution (pH 7.0). As shown in Fig. S5,† just little loss in the current signal was observed after 100 cycles. The sensor was stored dry at room temperature and measured at intervals of ten days. The sensor retained 96.3%, 90.6% and 85.9% of its initial amperometric responses after 10, 20 and 30 days as shown in Fig. S6,† which indicated that the sensor has long-term storage and is stable for electrochemical application.
3.6.4. Real sample analysis. In order to evaluate the analytical reliability and practical application of the proposed sensor, real blood serum sample was selected for analysis. The serum sample was diluted with 0.2 M PBS (pH 7.0) buffer solution by a factor of 1/10 (v/v) and then spiked with appropriate amount of DA. Real sample was analyzed by standard addition method, a linear relation was obtained for the catalytic current I (μA) versus DA concentration C (μM) (I = 1.504 + 1.166 × C, R2 = 0.998). Using this equation, the analytical results were summarized in Table 2. The recovery of the spiked samples ranged between 97.2% and 100.27%, which is acceptable within the allowed error range.
Table 2 Results of the recovery tests obtained for determination of DA in real sample
| Sample |
CDA/(μM) (expected) |
CDA/(μM) (recovered) |
Recovery (%) |
| 1 |
5.0 |
4.86 |
97.20 |
| 2 |
10.0 |
10.02 |
100.20 |
| 3 |
15.0 |
15.04 |
100.27 |
| 4 |
20.0 |
19.97 |
99.85 |
| 5 |
25.0 |
24.85 |
99.40 |
4. Conclusion
In this work, a composite film based on polyoxometalates, copper oxide and chitosan–palladium was fabricated. Fast electron transfer and high electrocatalytic activity towards the oxidation DA at the composite film were observed because of the synergistic effects among three active components. On this basis, a DA sensor with high selectivity was explored, free from UA and AA. The proposed sensor exhibited excellent electrochemical sensing performance such as wide linear range, low detection limit, high sensitivity as well as good reproducibility and long-term stability, which indicates the {PEI/[(P2W17V–CuO)/(CS–Pd)]7/(P2W17V–CuO)} composite film can be a potential candidate for routine dopamine analysis. This electrochemical sensor has the advantages of ease of preparation and low production cost.
Acknowledgements
This work was financially supported by the NSF of China (21371041, 51572063, 21406043), the Science and Technology Innovation Foundation of Harbin (2014RFXXJ076).
References
- Y. Y. Ling, Q. A. Huang, M. S. Zhu, D. X. Feng, X. Z. Li and Y. Wei, J. Electroanal. Chem., 2013, 693, 9–15 CrossRef CAS.
- S. F. Hou, M. L. Kasner, S. J. Su, K. Patel and R. Cuellari, J. Phys. Chem. C, 2010, 114, 14915–14921 CAS.
- Q. T. Huang, H. Q. Zhang, S. R. Hu, F. M. Li, W. Weng, J. H. Chen, Q. X. Wang, Y. S. He, W. X. Zhang and X. X. Bao, Biosens. Bioelectron., 2014, 52, 277–280 CrossRef CAS PubMed.
- L. I. Goldberg, Pharmacol. Rev., 1972, 24, 1–29 CAS.
- Z. H. Guo and S. J. Dong, Electroanalysis, 2005, 17, 607–612 CrossRef CAS.
- L. L. Li, H. Y. Liu, Y. Y. Shen, J. R. Zhang and J. J. Zhu, Anal. Chem., 2011, 83, 661–665 CrossRef CAS PubMed.
- A. Abbaspour, A. Khajehzadeh and A. Ghaffarinejad, Analyst, 2009, 134, 1692–1698 RSC.
- W. J. Barretoa, S. R. G. Barreto, R. A. Ando, P. S. Santos, E. DiMauro and T. Jorge, Spectrochim. Acta, Part A, 2008, 71, 1419–1424 CrossRef PubMed.
- J. L. Chen, X. P. Yan, K. Meng and S. F. Wang, Anal. Chem., 2011, 83, 8787–8793 CrossRef CAS PubMed.
- V. S. Y. Lin, C. Y. Lai, J. G. Huang, S. A. Song and S. Xu, J. Am. Chem. Soc., 2001, 123, 11510–11511 CrossRef CAS PubMed.
- S. R. Wallenborg, L. Nyholm and C. E. Lunte, Anal. Chem., 1999, 71, 544–549 CrossRef CAS PubMed.
- Y. H. Park, X. Zhang, S. S. Rubakhin and J. V. Sweedler, Anal. Chem., 1999, 71, 4997–5002 CrossRef CAS PubMed.
- V. Carrera, E. Sabater, E. Vilanova and M. A. Sogorb, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 847, 88–94 CrossRef CAS PubMed.
- C. Muzzi, E. Bertocci, L. Terzuoli, B. Porcelli, I. Ciari, R. Pagani and R. Guerranti, Biomed. Pharmacother., 2008, 62, 253–258 CrossRef CAS PubMed.
- M. Rafiee and L. Khalafi, Electrochim. Acta, 2010, 55, 1809–1813 CrossRef CAS.
- B. B. Li, Y. S. Zhou, W. Wu, M. Liu, S. R. Mei, Y. K. Zhou and T. Jing, Biosens. Bioelectron., 2015, 67, 121–128 CrossRef CAS PubMed.
- X. F. Liu, S. P. Wei, S. H. Chen, D. H. Yuan and W. Zhang, Appl. Biochem. Biotechnol., 2014, 173, 1717–1726 CrossRef CAS PubMed.
- X. X. Weng, Q. X. Cao, L. X. Liang, J. R. Chen, C. P. You, Y. M. Ruan, H. J. Lin and L. J. Wu, Talanta, 2013, 117, 359–365 CrossRef CAS PubMed.
- S. J. Li, J. Z. He, M. J. Zhang, R. X. Zhang, X. L. Lv, S. H. Li and H. Pang, Electrochim. Acta, 2013, 102, 58–65 CrossRef CAS.
- X. F. Liu, L. Zhang, S. P. Wei, S. H. Chen, X. Ou and Q. Y. Lu, Biosens. Bioelectron., 2014, 57, 232–238 CrossRef CAS PubMed.
- C. Q. Wang, J. Du, H. W. Wang, C. Zou, F. X. Jiang, P. Yang and Y. K. Du, Sens. Actuators, B, 2014, 204, 302–309 CrossRef CAS.
- Y. Y. Wu, L. L. Cui, Y. Liu, G. J. Lv, T. Pu, D. J. Liu and X. Q. He, Analyst, 2013, 138, 1204–1211 RSC.
- R. Doyle, C. B. Breslin and A. D. Rooney, Chem. Biochem. Eng. Q., 2009, 23, 93–98 CAS.
- S. Pourbeyram, Sens. Actuators, B, 2014, 192, 105–110 CrossRef CAS.
- L. H. Gao, J. F. Zhang, H. L. Wang, X. Y. Lin, J. M. Qi and K. Z. Wang, Electrochim. Acta, 2015, 166, 215–222 CrossRef CAS.
- Y. Sahraoui, A. Sbartai, S. Chaliaa, A. Maaref, A. Haddad and N. Jaffrezic-Renault, Electroanalysis, 2015, 27, 1–10 CrossRef.
- X. L. Chen, B. Souvanhthong, H. Wang, H. W. Zheng, X. H. Wang and M. X. Huo, Appl. Catal., B, 2013, 138, 161–166 CrossRef.
- H. L. Lv, W. W. Guo, K. F. Wu, Z. Y. Chen, J. Bacsa, D. G. Musaev, Y. V. Geletii, S. M. Lauinger, T. Q. Lian and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 14015–14018 CrossRef CAS PubMed.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed.
- S. P. Liu, L. Xu, F. Y. Li, W. H. Guo, Y. Xing and Z. X. Sun, Electrochim. Acta, 2011, 56, 8156–8162 CrossRef CAS.
- B. Keita, R. Contant, P. Mialane, F. Sécheresse, P. de Oliveiraa and L. Nadjo, Electrochem. Commun., 2006, 8, 767–772 CrossRef CAS.
- C. L. Zhou, S. Li, W. Zhu, H. J. Pang and H. Y. Ma, Electrochim. Acta, 2013, 113, 454–463 CrossRef CAS.
- D. M. Fernandes and C. Freire, ChemElectroChem, 2015, 2, 269–279 CrossRef CAS.
- W. H. Guo, X. L. Tong and S. B. Liu, Electrochim. Acta, 2015, 173, 540–550 CrossRef CAS.
- S. Reddy, B. E. K. Swamy and H. Jayadevappa, Electrochim. Acta, 2012, 61, 78–86 CrossRef CAS.
- J. H. Li, J. L. Liu, G. R. Tan, J. B. Jiang, S. J. Peng, M. Deng, D. Qian, Y. L. Feng and Y. C. Liu, Biosens. Bioelectron., 2014, 54, 468–475 CrossRef CAS PubMed.
- X. M. Chen, Z. X. Cai, Z. Y. Huang, M. Oyama, Y. Q. Jiang and X. Chen, Electrochim. Acta, 2013, 97, 398–403 CrossRef CAS.
- S. Khadempir, A. Ahmadpour, M. T. H. Mosavian, N. Ashraf, F. F. Bamoharram, S. G. Mitchell and J. M. de la Fuente, RSC Adv., 2015, 5, 24319–24326 RSC.
- F. C. Yang, J. Wang, Y. Cao, L. Zhang and X. Zhang, Sens. Actuators, B, 2014, 205, 20–25 CrossRef CAS.
- Y. E. Miao, S. X. He, Y. L. Zhong, Z. Yang, W. W. Tjiu and T. X. Liu, Electrochim. Acta, 2013, 99, 117–123 CrossRef CAS.
- M. Najafi, M. A. Khalilzadeh and H. Karimi-Maleh, Food Chem., 2014, 158, 125–131 CrossRef CAS PubMed.
- J. A. Farmer and C. T. Campbell, Science, 2010, 329, 933–936 CrossRef CAS PubMed.
- X. M. Chen, G. H. Wu, J. M. Chen, X. Chen, Z. X. Xie and X. R. Wang, J. Am. Chem. Soc., 2011, 133, 3693–3695 CrossRef CAS PubMed.
- R. J. White, R. Luque, V. L. Budarin, J. H. Clark and D. J. Macquarrie, Chem. Soc. Rev., 2009, 38, 481–494 RSC.
- H. P. Liang, T. G. J. Jones, N. S. Lawrence, L. Jiang and J. S. Barnard, J. Phys. Chem. C, 2008, 112, 4327–4332 CAS.
- H. Huang, N. F. Hu, Y. H. Zeng and G. Zhou, Anal. Biochem., 2002, 308, 141–151 CrossRef CAS PubMed.
- E. Sin, S. S. Yi and Y. S. Lee, J. Mol. Catal. A: Chem., 2010, 315, 99–104 CrossRef CAS.
- M. Adlim, M. A. Bakar, K. Y. Liew and J. Ismail, J. Mol. Catal. A: Chem., 2004, 212, 141–149 CrossRef CAS.
- N. V. Kramareva, A. Y. Stakheev, O. P. Tkachenko, K. V. Klementiev, W. Grünert, E. D. Finashina and L. M. Kustov, J. Mol. Catal. A: Chem., 2004, 209, 97–106 CrossRef CAS.
- Y. X. Sun, Y. Guo, Q. Z. Lu, X. L. Meng, W. X. Hua, Y. L. Guo, Y. S. Wang, X. H. Liu and Z. G. Zhang, Catal. Lett., 2005, 100, 213–217 CrossRef CAS.
- M. Abbessi, R. Contant, R. Thouvenot and G. Hervé, Inorg. Chem., 1991, 30, 1695–1702 CrossRef CAS.
- R. Ahmad, M. Vaseem, N. Tripathy and Y. B. Hahn, Anal. Chem., 2013, 85, 10448–10454 CrossRef CAS PubMed.
- H. Huang, Q. Yuan and X. R. Yang, Colloids Surf., B, 2004, 39, 31–37 CrossRef CAS PubMed.
- L. Kang, H. Y. Ma, Y. Yu, H. J. Pang, Y. B. Song and D. Zhang, Sens. Actuators, B, 2013, 177, 270–278 CrossRef CAS.
- Y. Y. Bao, L. H. Bi and L. X. Wu, J. Solid State Chem., 2011, 184, 546–556 CrossRef CAS.
- M. M. Guo, P. S. Wang, C. H. Zhou, Y. Xia, W. Huang and Z. L. Li, Sens. Actuators, B, 2014, 203, 388–395 CrossRef CAS.
- M. Li, X. J. Bo, Y. F. Zhang, C. Han and L. P. Guo, Biosens. Bioelectron., 2014, 56, 223–230 CrossRef CAS PubMed.
- A. T. Kuvarega, R. W. M. Krause and B. B. Mamba, J. Phys. Chem. C, 2011, 115, 22110–22120 CAS.
- D. Zhang, H. Y. Ma, Y. Y. Chen, H. J. Pang and Y. Yu, Anal. Chim. Acta, 2013, 792, 35–44 CrossRef CAS PubMed.
- B. Keita, F. Girard, L. Nadjo, R. Contant, J. Canny and M. Richet, J. Electroanal. Chem., 1999, 478, 76–82 CrossRef CAS.
- B. Keita, I. M. Mbomekalle, L. Nadjo, P. de Oliveira, A. Ranjbari and R. Contant, C. R. Chim., 2005, 8, 1057–1066 CrossRef CAS.
- S. Q. Liu, D. G. Kurth, B. Bredenkotter and D. Volkmer, J. Am. Chem. Soc., 2002, 124, 12279–12287 CrossRef CAS PubMed.
- X. L. Wang, Q. Gao, A. X. Tian, H. L. Hu and G. C. Liu, J. Solid State Chem., 2012, 187, 219–224 CrossRef CAS.
- M. H. Huang, L. H. Bi, Y. Shen, B. F. Liu and S. J. Dong, J. Phys. Chem. B, 2004, 108, 9780–9786 CrossRef CAS.
- L. L. Cui, T. Pu, Y. Liu and X. Q. He, Electrochim. Acta, 2013, 88, 559–564 CrossRef CAS.
- E. Sabatani and I. Rubinstein, J. Phys. Chem., 1987, 91, 6663–6669 CrossRef CAS.
- L. Chen, L. Tian, L. Liu, X. F. Tian, W. B. Song, H. D. Xu and X. H. Wang, Sens. Actuators, B, 2005, 110, 271–278 CrossRef CAS.
- M. Ammam, I. M. Mbomekalle, B. Keita, L. Nadjo and J. Fransaer, Electrochim. Acta, 2010, 55, 3118–3122 CrossRef CAS.
- B. Keita, A. Belhouari, L. Nadjo and R. Contant, J. Electroanal. Chem., 1995, 381, 243–250 CrossRef.
- G. M. Jiang, X. F. Gu, G. Q. Jiang, T. T. Chen, W. Y. Zhan and S. Tian, Sens. Actuators, B, 2015, 209, 122–130 CrossRef CAS.
- F. Gao, X. L. Cai, X. Wang, C. Gao, S. L. Liu, F. Gao and Q. X. Wang, Sens. Actuators, B, 2013, 186, 380–387 CrossRef CAS.
- S. Palanisamy, S. Ku and S. M. Chen, Microchim. Acta, 2013, 180, 1037–1042 CrossRef CAS.
- S. Reddy, B. E. Kumara Swamy and H. Jayadevapp, Electrochim. Acta, 2012, 61, 78–86 CrossRef CAS.
- L. Q. Lin, J. H. Chen, H. Yao, Y. Z. Chen, Y. J. Zheng and X. H. Lin, Bioelectrochemistry, 2008, 73, 11–17 CrossRef CAS PubMed.
- M. Mallesha, R. Manjunatha, C. Nethravathi, G. S. Suresh, M. Rajamathi, J. S. Melo and T. V. Venkatesha, Bioelectrochemistry, 2011, 81, 104–108 CrossRef CAS PubMed.
- R. W. Chen, Y. Z. Wang, Y. Liu and J. H. Li, RSC Adv., 2015, 5, 85065–85072 RSC.
- X. X. Liu, H. Zhua and X. R. Yang, RSC Adv., 2014, 4, 3706–3712 CAS.
- J. Zhao, Y. H. Yu, B. Weng, W. M. Zhang, A. T. Harris, A. I. Minett, Z. L. Yue, X. F. Huang and J. Chen, Electrochem. Commun., 2013, 37, 32–35 CrossRef CAS.
- D. B. Gorle and M. A. Kulandainathan, RSC Adv., 2016, 6, 19982–19991 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra09819c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.