
Variable temperatures 1H NMR technique on the kinetics and thermodynamic parameters of equilibrium between the Z and E isomers in a stable phosphorus ylide involving an imidazole
Abstract
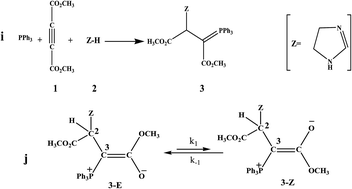
Analysis of 1H NMR spectra of a special ylide involving an imidazole at variable temperatures has been employed to study the kinetics of the interchangeable process in the equilibrium between the two Z- and E-isomers of a stable phosphorus ylide. Activation data (k−1, k1, ktotal, Ea1, Ea−1, Eatotal, ΔH‡, ΔG‡, ΔS‡) and thermodynamic parameters (Ke, ΔH°, ΔG° and ΔS°) for the forward and reverse steps of the opposing reaction were obtained by means of the variable temperature 1H NMR technique and T-jump method. The 1H NMR results indicate that the interchangeable process of rotational isomers follows first-order kinetics with respect to both the forward and reverse reactions. The process was also entropy controlled.
 Please wait while we load your content...
Please wait while we load your content...

