
Heterogeneous photooxidation of sulfur dioxide in the presence of airborne mineral dust particles†
Abstract
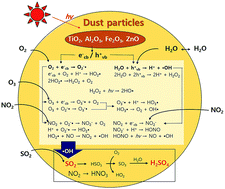
The photocatalytic effect of Arizona Test Dust (ATD) particles on SO2 oxidation was investigated in both the absence and the presence of atmospheric oxidants (e.g., NOx and O3) under varying relative humidity (RH) using a 2 m3 indoor photo-irradiation chamber. To the best of our knowledge, this is the first time that the kinetic rate constant has been determined for heterogeneous photooxidation of SO2 in the presence of airborne mineral dust particles. Chamber-generated sulfate data, measured using a particle into liquid sampler coupled with ion chromatography, were used to estimate the kinetic uptake coefficient (γSO42−) of SO2 corresponding to sulfate production on ATD particles. The γSO42− value (γSO42−,dark) in the presence of UV light was significantly higher than that (γSO42−,dark) obtained in the dark. For example, γSO42−,light was (1.15 ± 0.17) × 10−5 and γSO42−,dark was (1.16 ± 0.11) × 10−6 at 20% RH. Both γSO42−,light and γSO42−,dark values increased exponentially as RH increased from 20% to 81%. The heterogeneous photocatalytic oxidation of SO2 was also considerably enhanced by both O3 and NO2. To characterize the reaction between sulfuric acid and inorganic species of dust particles, aerosol acidity ([H+]C-RUV, μmol L−1 by aerosol volume) was measured using colorimetry integrated with a reflectance UV-Visible spectrometer. Under the experimental conditions employed (sulfate : dust particle = 1 : 20 in mass), the value determined for [H+]C-RUV was much lower than the value of [H+] determined from the concentrations of sulfate and ammonium ions measured using ion chromatography, suggesting that most sulfuric acid reacted with alkaline components of ATD particles.
 Please wait while we load your content...
Please wait while we load your content...

