
The catalytic characteristics of NocB in nocathiacin biosynthesis from Nocardia sp. ATCC 202099
Xuri Wu†
,
Peiyu Huang†,
Yanjiu Xue,
Weiying Liu,
Min Ma and
Yijun Chen*
State Key Laboratory of Natural Medicines and Laboratory of Chemical Biology, China Pharmaceutical University, 24 Tongjia St., Nanjing, Jiangsu Province 210009, People's Republic of China. E-mail: yjchen@cpu.edu.cn; Fax: +86-25-83271031; Tel: +86-25-83271045
First published on 26th July 2016
Abstract
Nosiheptide and nocathiacin are thiopeptides within the same series with high similarity from chemical structure to organization of the biosynthetic gene cluster. Previously, NosB, a cytochrome P450-like enzyme, was demonstrated to be responsible for the hydroxylation of Glu-6 during nosiheptide maturation. Based on 64% amino acid sequence identity, NocB from Nocardia sp. ATCC 202099 is expected to exhibit a similar catalytic function to NosB. After replacing nosB by nocB in nosiheptide-producing Streptomyces actuosus ATCC 25421, NocB was proved to be a cytochrome P450-like monooxygenase responsible for the hydroxylation of Glu-6 at the γ-position in the biosyntheses of both nosiheptide and nocathiacin. Enzyme kinetics and structural difference between nosiheptide and nocathiacin further revealed that NocB is less active than NosB towards unglycosylated intermediate 3 containing a bicyclic core structure.
Introduction
Thiopeptide antibiotics are a family of sulfur-rich, highly modified natural products and their biosynthetic pathways have been found to go through ribosomally synthesized and post-translationally modified peptide (RiPPs) systems.1,2 The attractive biological activities possessed by characterized members of the thiopeptide family have led to extensive investigations focusing on the elucidation of the molecular mechanisms of their biosynthesis. Nosiheptide is produced by S. actuosus ATCC 25421 and contains a bicyclic core in the structure.3 As another member of thiopeptide in the same series, nocathiacin is produced by Nocardia sp. ATCC 202099 with a similar structure to unglycosylated nosiheptide (Fig. 1A). However, in addition to the glycosylation and other modifications, nocathiacin features a characteristic difference, in which a tricyclic core is formed by a rigid constraint through indole ring via an ether bond and two ester linkages.4 Given the fact that the biosynthetic gene clusters of nosiheptide and nocathiacin were respectively identified in 2009 and 2010,5,6 comparative analysis of these two clusters has become possible. Except for glycosylation as the final step to produce mature nocathiacin, high homology of the biosynthetic genes has suggested them to play similar roles in the formation of the core structure (Fig. 1B).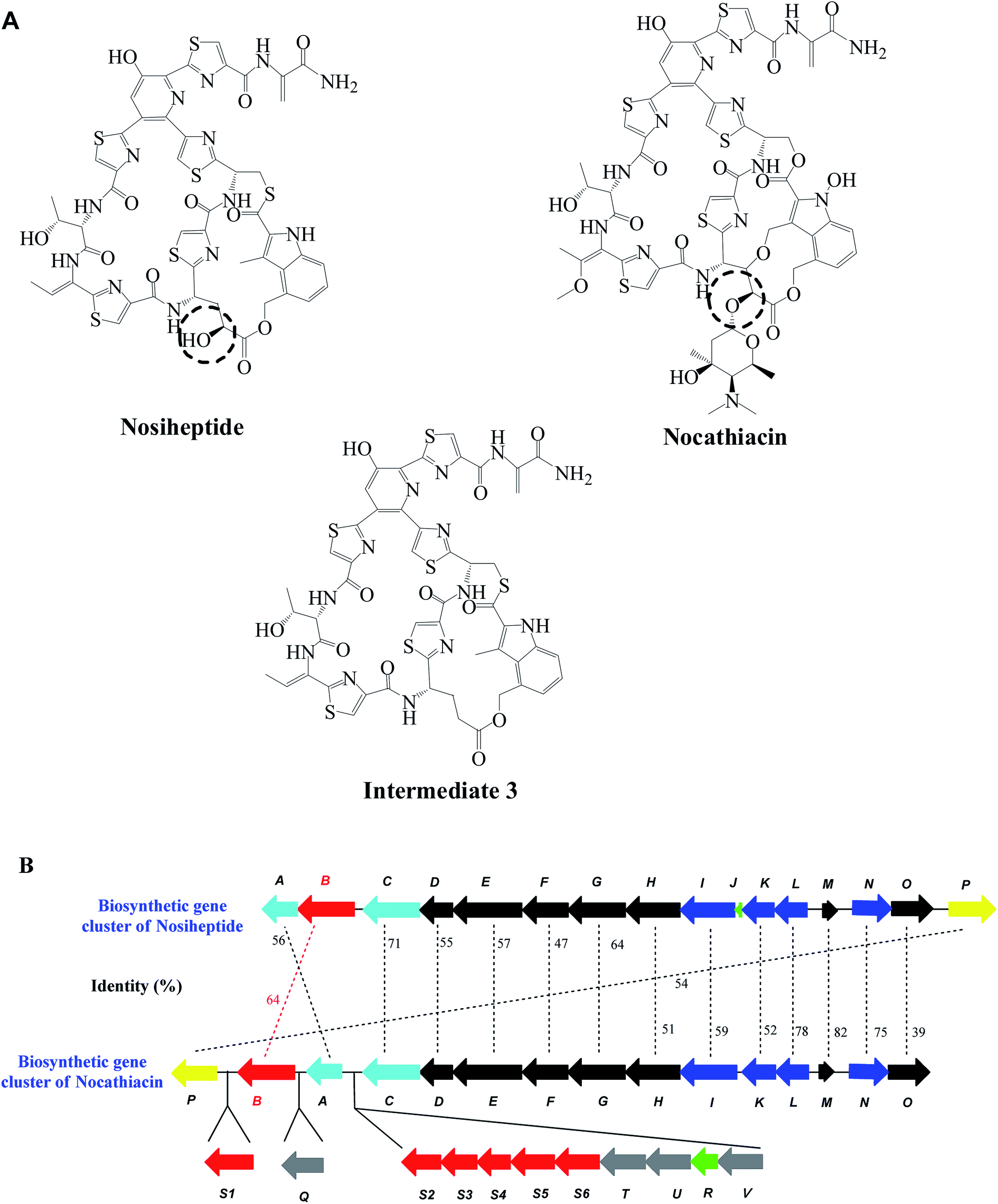 | ||
| Fig. 1 Structures and partial gene cluster comparison between nosiheptide and nocathiacin. (A) Structures of nosiheptide and nocathiacin. The hydroxylation sites of Glu-6 are shown in dotted circles, respectively. Intermediate 3 was produced after the deletion of nosB in S. actuosus ATCC 25421. (B) Organization and amino acid sequence comparison of NosB and NocB. | ||
We recently demonstrated the late-stage tailoring of nosiheptide by genetic manipulation in its producing strain.7 The intermediate structure produced by NosB-inactivated mutant indicated that NosB is responsible for the hydroxylation of Glu-6 at its γ-position, which was further confirmed by enzymatic catalysis in vitro. Meanwhile, NosC was proven to hydroxylate the Pyr-3 position both in vivo and in vitro. However, the intermediate generated from NosC-inactivated mutant contained an extra bis-dehydroalanine (Dha) tail, originating from the absence of a functional NosA, an enzyme responsible for the final enamine dealkylation.8 To verify whether the inactivation of NosC completely prevented enamine dealkylation by NosA, two mutants, ΔnosAB and ΔnosBC were constructed and the structures of the intermediates were characterized. The differences on structure and oxidative state demonstrated that NosB and NosC are cytochrome P450-like enzymes and the hydroxylation at Pyr-3 position catalyzed by NosC should occur before the cleavage of the bis-Dha tail by NosA, whereas NosB deletion had no effects on NosA activity. Therefore, three oxidative routes for the maturation of nosiheptide were proposed,9 in which the substrate specificity of NosB is relatively broad.
After the inspection of the biosynthetic gene cluster of nocathiacin, we found that NocB, a analogous protein to NosB, exhibits three major characteristics: a conserved threonine residue to involve in O2 activation, a conserved EXXR motif and a heme-binding domain GXXXCXG, which are typical for cytochrome P450 enzymes.10 Nucleotide sequence analysis also indicated that NocB shares 64% identity to NosB (Fig. 1B),6 suggesting that NocB might be a cytochrome P450-like enzyme and belongs to the same subfamily as NosB to possess similar function.9,11 On the other hand, the differences between nocathiacin and nosiheptide in structure and gene cluster organization6 do raise a question on whether these two enzymes from different gene clusters have the same substrate specificity and catalytic properties. Unfortunately, given the difficulty on genetic manipulation in Nocardia sp., it is impossible to directly probe the function and substrate specificity of NocB in the native host. To properly address this question and gain further insights into the biosynthetic machinery, we heterogeneously replaced NosB by NocB to determine the production of mature nosiheptide and compared their catalytic characteristics in the present study. Our results demonstrated that NocB is a cytochrome P450-like enzyme responsible for the hydroxylation of the γ-position of Glu-6 in nosiheptide precursor and its catalytic activity is lower than that of NosB towards unglycosylated intermediate 3 containing a bicyclic core structure.
Results and discussion
Construction of mutant L1307 with nocB gene substitution
Previously, a nosB deletion strain Streptomyces actuosus L1120 was obtained by gene knockout and NosB was characterized as a cytochrome P450-like enzyme involving in the hydroxylation of the γ-position of Glu-6 in the biosynthesis of nosiheptide.9 However, the function of NocB, a homologous protein of NosB, has not been able to experimentally confirm due to the difficulty on genetic manipulation in the native host, the nocathiacin-producing Nocardia strain.6,12 Given that nocathiacin biosynthesis shares a common paradigm for forming the characteristic thiopeptide core via a route mechanistically similar to that of nosiheptide,6,13,14 the biosynthetic gene cluster of nosiheptide may be able to tolerate the exchange of noc genes to produce nosiheptide. Consequently, the function of NocB could be investigated by replacing NosB in nosiheptide-producing Streptomyces strain. As shown in Fig. 2A, nocB gene with its native start codon (ATG) was amplified and introduced into S. actuosus L1120 to generate a nocB+ strain (designated as S. actuosus L1307) without addition of exogenous promoter(s). The genotype of L1307 mutant strain was further confirmed by bacterial liquid PCR (Fig. 2B). The target fragments were recovered and further confirmed by DNA sequencing. As a result, the mutant of nosiheptide-producing strain L1307, in which nosB gene was replaced by nocB, was successfully constructed and used for subsequent experiments. | ||
| Fig. 2 Schematic representation of the method used for nocB gene replacement. (A) Homologous double crossover between the plasmid containing nocB gene and the chromosome of S. actuosus L1120 generating nocB+ variant; LF and RF represent the homologous fragments flanking nosB gene, and aac(3) IV represents the apramycin resistance gene. (B) Identification of nocB+ and nosB− mutants by PCR using primers nosAB-S2F and nosAB-S2R. M, DNA ladders; lane 1, nocB+ (L1307 mutant). The expected size of positive fragment containing nocB gene is 2265 bp. Lane 2, nosB− (L1120 mutant). The expected fragment without nosB gene is 1159 bp. | ||
Validation of catalytic activity for NocB in vivo
To detect nosiheptide production in S. actuosus L1307 mutant with NocB substitution, the mutant strain was fermented for 4 days at 32 °C, and the cultures were extracted and analyzed by HPLC. As shown in Fig. 3A, L1307 mutant strain could produce nosiheptide at 15.1 ± 0.6 mg L−1, about a quarter of that produced by WT strain (64.9 ± 2.1 mg L−1). These data suggested that NocB is indeed a cytochrome P450-like enzyme with similar function as NosB,9 but the catalytic efficiency of NocB might be lower than that of NosB. Meanwhile, a new intermediate appeared and accumulated in the mutant strain L1307 (Fig. 3B). Based on the retention time on HPLC and molecular mass determined by LC-MS/MS (Fig. 3B and C), the structural identity of this new intermediate was revealed to be the same as the intermediate (3) from nosB deletion strain S. actuosus L1120 (Fig. 1A). The production of intermediate 3 in L1307 mutant strain was calculated to be 51.8 ± 3.1 mg L−1 according to the authentic sample. After data analysis, we found that the average values of combined nosiheptide and intermediate 3 in L1307 mutant strain was close to the total amount of nosiheptide in WT. Therefore, the transcription and translation levels of partial nosiheptide gene cluster, the compatibility of nocB with the cluster and the kinetic characteristics of NocB were systematically investigated to explore the key factors to affect nosiheptide production catalyzed by NocB. | ||
| Fig. 3 Production of nosiheptide and intermediate 3 in S. actuosus ATCC 25421 and L1307 mutant. (A) The production of nosiheptide and intermediate 3 in S. actuosus ATCC 25421 and L1307 mutant. Data are averages from 3 independent experiments with standard deviations. (B) HPLC analyses of the metabolites from the cultures. (i) S. actuosus ATCC 25421; (ii) S. actuosus L1307 mutant; (iii) purified intermediate from S. actuosus L1120 mutant. (C) HRMS of the intermediate 3 from S. actuosus L1307 mutant. | ||
Effects of gene transcription on nosiheptide biosynthesis
In nosiheptide-producing S. actuosus ATCC 25421, the maturation of final product nosiheptide relies on the coordination of NosA, NosB and NosC. The incompatibility of any of these genes with the host and the decrease of catalytic activity of any enzymes would result in incomplete maturation of nosiheptide and accumulation of corresponding intermediate(s).9 Within the biosynthetic gene cluster of nosiheptide, there is a series of regulatory elements to accurately coordinate nosiheptide biosynthesis. The substitution of nosB by nocB might interfere the endogenous regulatory system to influence the biosynthetic machinery for nosiheptide maturation. Therefore, we compared the transcription levels of nocB in S. actuosus L1307 and nosB in wild type (WT) to evaluate the compatibility between nocB gene and nosiheptide gene cluster in the mutant host. Total RNAs extracted from 2 day fermentation cultures were analyzed by RT-qPCR. Relative transcription levels of nocB in L1307 and nosB in WT were determined according to the 2−ΔΔt method. As shown in Fig. 4A, using the transcription level of nosB in WT as a background value (defined as 1.0 ± 0.1), the transcription level of nocB in L1307 mutant was 1.36 ± 0.3 (P < 0.05), indicating that the transcription of nocB in Streptomyces strain can actually work well under the exogenous regulatory control. | ||
| Fig. 4 Relative transcription of nosA and noc(s)B genes in S. actuosus ATCC 25421 and L1307 mutant. (A) Relative transcription levels of noc(s)B gene in S. actuosus L1307 mutant and WT. (B) Relative transcription levels of nosA gene in S. actuosus L1307 mutant and WT. Data are averages of 3 independent experiments with standard deviations (*, P < 0.05). | ||
nosA is located at the downstream of nocB in the biosynthetic gene cluster of nosiheptide (Fig. 1B), suggesting that nocB substitution in the gene cluster may also affect the transcription of nosA. Therefore, we compared the transcription levels of nosA between mutant L1307 and WT. After replacing nosB by nocB, the level of nosA transcription in L1307 mutant strain surprisingly increased approximately 22-fold (Fig. 4B), which ensures sufficient quantity of NosA to be expressed for final maturation of nosiheptide.8,9 Therefore, the efficient transcription of nocB and nosA in S. actuosus L1307 unambiguously ruled out the possibility of incomplete maturation of nosiheptide and accumulation of intermediate 3 due to decreased transcription of nocB and nosA.
Heterogeneous protein expression in E. coli to assess potential impact on nosiheptide biosynthesis
Although nocB substitution did not influence the transcription of nocB and its downstream nosA gene in Streptomyces strain, the expression compatibility of exogenous nocB could still be affected by the gene replacement. Typically, constitutive PermE* promoter is used to detect heterologous expression of proteins in Streptomyces strains.15 However, in the case of nocB, this promoter resulted in extremely low expression (data not shown) due possibly to the insertion at attB site in the chromosome of nosB-deletion strain. Based on our previous study, NosB and NosC, two similar cytochrome P450-like enzymes with high homology to NocB, could be functionally expressed well in E. coli system.9 Alternatively, we investigated and compared the expression compatibility of nocB in L1307 mutant and nosB in WT using a fluorescence reporter in a heterogeneous system with E. coli cells, in which protein expressions were controlled under the same promoter to make a comparison. According to the scheme shown in Fig. 5A, a 2459 bp gene fragment containing full length of nosC gene and nocB gene (lacking the stop codon TGA at its C-terminus) and green fluorescent protein (gfp) gene fragment were ligated into pET28a(+) to generate an expression plasmid pET28a-L1307-c-nocb-gfp, in which gfp gene was used as a reporter for nocB. Similarly, pET28a-WT-c-nosb-gfp was constructed and used to comparatively indicate the expression of NosB. | ||
| Fig. 5 Relative expression of NosA and Nos(c)B in E. coli. (A) Construction of fusion proteins with green fluorescent protein (GFP). (B) Relative expression levels of Nos(c)B by detection of fluorescence intensity. (C) Relative expression levels of NosA by detection of the fluorescence intensity. Data are averages of 3 independent experiments with standard deviations (*, P < 0.005). | ||
After IPTG induction in E. coli strain BL21 (DE3), the cell extracts were used to detect the fluorescence intensity from GFP. As shown in Fig. 5B, when the fluorescence intensity of NosB-GFP from WT was defined as 100%, relative fluorescence intensity (RFI) of NocB-GFP was 284%, suggesting that NocB could be functionally and sufficiently expressed and be compatible with NosC in E. coli system. To investigate whether nocB substitution affects the translation of downstream gene nosA, we thus constructed two nosA-gfp reporters, WT-CBA-GFP and L1307-CBA-GFP. When the fluorescence intensity of NosA-GFP from WT was defined as 100%, RFI of NosA-GFP from L1307 mutant was 198%, revealing that nocB actually increased the expression of the downstream gene nosA under the same experimental conditions in E. coli (Fig. 5C). The results suggested that NocB from Nocardia sp. might be able to compatibly express within nosiheptide gene cluster in S. actuosus ATCC 25421, and a negative impact from protein expression on nosiheptide biosynthesis would be insignificant. Although direct comparison on protein expression in native Streptomyces host under the control of its native promoter would be desirable to truly reflect the influence on nosiheptide biosynthesis, the present heterogeneous expression in E. coli could still be served as an indicator to assess possible factors potentially affecting nosiheptide biosynthesis in Streptomyces.
Confirmation of the catalytic activity of NocB
Given that nocB is compatible with nosiheptide gene cluster at transcription and translation levels in the host strain, the lower production of nosiheptide in mutant L1307 could be due to lower catalytic activity of NocB against nosiheptide-related substrates. Thus, NocB was expressed in soluble form with a His6-tag at the N-terminus in E. coli, and purified to homogeneity by chromatographic steps of Ni-Sepharose and Q-Sepharose (Fig. 6). The enzymatic reaction by purified NocB was performed using intermediate 3 as a substrate according to that of NosB in our previous work.9 After the reaction and HPLC analysis, the product was identified to be nosiheptide, indicating that NocB indeed is a cytochrome P450-like enzyme with similar function as NosB, and most likely responsible for the hydroxylation of the γ-position of Glu-6 in nocathiacin biosynthesis. However, the residual substrate also suggested that the catalytic efficiency of NocB is lower than that of NosB when intermediate 3 is served as a substrate. | ||
| Fig. 6 SDS-PAGE for the purification of recombinant NocB and NosB. SDS-PAGE was performed on a 12% gel under reducing condition. M, molecular weight markers; lane 1, expression of NosB in cell-free extracts of E. coli BL21 containing pET28a-nosB; lane 2, purified recombinant NosB; lane 3, expression of NocB in cell-free extracts of E. coli BL21 containing pET28a-nocB; lane 4, purified recombinant NocB. | ||
Catalytic characteristics and substrate specificity of NocB
According to the analyses of transcription and translation of nocB, we found that nocB displays compatibility with its host L1307 to produce more mRNA and protein in E. coli-based model system than that of WT S. actuosus. Hence, the reduced production of nosiheptide and accumulation of intermediate 3 should attribute to intrinsic catalytic properties of NocB, especially its substrate specificity. The relative kinetic parameters for NocB and NosB in the hydroxylation of intermediate 3 were determined and compared with substrate concentrations varied from 41.5 μM to 165.8 μM due to lower aqueous solubility of intermediate 3. The apparent kinetic constants were obtained by analyzing the experimental data with reciprocal Lineweaver–Burk method. As shown in Table 1, apparent Km value for NocB was approximately 2.8-fold greater than that of NosB towards intermediate 3, whereas apparent catalytic efficiency (kcat/Km) of NosB was 19.8-fold higher than that of NocB. Although the kinetic data were relative and apparent within the range of substrate concentration used in the experiments, they could be used to make a reasonable comparison. These data indicated that NocB is not an efficient enzyme to hydroxylate Glu-6 at the γ-position of intermediate 3 compared to its native enzyme NosB.| Enzyme | Apparent Km (mM) | Apparent Vmax (μmol min−1 mg−1) | Apparent kcat (s−1) | Apparent kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| a The apparent kinetic constants were determined with intermediate 3 as a substrate, and calculated based on the Michaelis–Menten model. All experiments were repeated three times. | ||||
| NocB | 0.61 | 3.06 | 2.23 | 3.66 × 103 |
| NosB | 0.22 | 19.37 | 15.95 | 7.25 × 104 |
Even though both nosiheptide and nocathiacin are members of e series of thiopeptides with similar chemical structure,16–18 nosiheptide contains a bicyclic core, whereas the central structure of nocathiacin is tricyclic. Accordingly, the native substrate of NocB during nocathiacin biosynthesis should be an unglycosylated, tricyclic intermediate whose Glu-6 position was not hydroxylated.6,9 Compared to intermediate 3, the extra ether bond and two ester linkages constructing the third cyclic ring in the native substrate for NocB makes the ring system more rigid and less flexible. This rigid tricyclic intermediate may fit better with the active site of NocB than intermediate 3, thereby yielding low efficiency of NocB.
Conclusions
In summary, we have characterized the function of NocB as a cytochrome P450-like monooxygenase by heterologous complementation and enzymatic reaction, which hydroxylates Glu-6 at the γ-position in the biosyntheses of nosiheptide and nocathiacin. The compatibility of NocB with its host was proven by transcriptional and translational analyses, and the hydroxylation site by NocB in the biosynthesis of nosiheptide was also confirmed. In addition, the accumulation of a non-hydroxylated intermediate in the nocB-substituted strain and relative kinetic parameters of NocB in the hydroxylation of intermediate 3 indicated that NocB is less active than NosB towards the bicyclic intermediate 3, and it may favor to catalyze the native tricyclic substrate with a rigid core structure.Experimental part
Materials
Details of bacterial strains and plasmids used in this study are given in Table 2. All DNA polymerases, restriction enzymes, PrimeScript™ RT reagent kit with gDNA Eraser (Perfect Real Time) and SYBR® Premix Ex Taq™ GC (Perfect Real Time) were purchased from TaKaRa (Dalian, China) or Fermentas (Thermo Fisher, USA). Glucose-6-phosphate dehydrogenase, ferredoxin–NADP reductase, ferredoxin and D-glucose-6-phosphate disodium salt were obtained from Sigma (USA). All chemicals, biochemicals and media used in this study were of analytical grade.| Strains or plasmids | Characteristic(s) | Reference |
|---|---|---|
| E. coli strains | ||
| ET12567/pUZ8002 | Donor strain for conjugation between E. coli and Streptomyces | Our laboratory |
| DH5α | Host for general cloning | TIANGEN |
| BL21 (DE3) | Host for protein expression | Novagen |
| WT-CB-GFP | Derivative of BL21 (DE3) containing nosC and nosB-gfp expression vector | This study |
| WT-CBA-GFP | Derivative of BL21 (DE3) containing nosC, nosB and nosA-gfp expression vector | This study |
| L1307-CB-GFP | Derivative of BL21 (DE3) containing nosC and nocB-gfp expression vector | This study |
| L1307-CBA-GFP | Derivative of BL21 (DE3) containing nosC, nocB and nosA-gfp expression vector | This study |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| Streptomyces actuosus | ||
| ATCC 25421 | Wild type strain, nosiheptide producing | ATCC |
| L1120 | nosB− | This study |
| L1307 | nocB+, nosB− | This study |
|
||
| Plasmids | ||
| pKC1139 | E. coli–Streptomyces shuttle vector, temperature sensitive replication in Streptomyces | Our laboratory |
| pMD19T simple vector | E. coli subcloning vector | TaKaRa |
| pET28a | E. coli protein expression vector | Novagen |
| pETDute-gfp | pETDute derivative containing green fluorescent protein gene (gfp) | Our laboratory |
| pKL1307 | pKC1139 derivative containing nocB gene and its homology arms, used for nocB replacement | This study |
| pET28a-WT-c-nosb-gfp | pET28a derivative containing nosC and nosB-gfp genes under the control of T7 promoter | This study |
| pET28a-WT-c-nosb-a-gfp | pET28a derivative containing nosC, nosB and nosA-gfp genes under the control of T7 promoter | This study |
| pET28a-L1307-c-nocb-gfp | pET28a derivative containing nosC and nocB-gfp genes under the control of T7 promoter | This study |
| pET28a-L1307-c-nocb-a-gfp | pET28a derivative containing nosC, nocB and nosA-gfp genes under the control of T7 promoter | This study |
DNA isolation and genetic manipulation
DNA isolation and manipulation in E. coli and Streptomyces were performed according to reported procedures.19 E. coli DH5α (Tiangen Biotech Co., Ltd., Beijing, China) was used as a host for plasmid transformations by CaCl2 method. Introducing plasmids into E. coli ET12567 for homologous recombination were carried out by electroporation in sterile cuvettes (2 mm electrode gap, Bio-Rad). The critical parameters evaluated for electro-transformation are as follows: voltage, 2.5 kV cm−1; capacitance, 25 μF; resistance, 200 Ω; time length, 5 ms.Construction of nocB-substitution mutant
nocB-substitution mutant was constructed according to our previous experimental procedure.7,9 A general PCR procedure was used to prepare the homologous fragments and the target gene fragments. Namely, a 50 μL PCR reaction mixture containing Prime STAR Max Premix (25 μL, 2×), PCR primers (1.0 μL, 20 mM) each, genomic DNA (1 μL, ca. 0.2 μg) and DMSO (2.5 μL) was prepared freshly, and the amplification condition was as follows: 96 °C for 10 s, 72 °C for 1.0 min (30 cycles); 72 °C for 5 min (1 cycle). The nocB gene was amplified from genomic DNA of Nocardia sp. ATCC 202099 using primers 1307F (5′-GGTGTTCGGTCATCGCAGGACCGCCTTCAGGAAGGCCCAGGTGGT-3′) and 1307R (5′-GTGGAGGCCCCCATGACCCGCGCCGACGCCGCGACCTACCCGTT-3′) to produce a fusion fragment LF-nocB. The left homologous fragment (LF) of nocB was amplified from genomic DNA of S. actuosus ATCC 25421 by PCR using primers 1306F (5′-AAGCTTCCCACGCGGTCGGGTGTGTCGAATCGTGCGGA-3′) and 1306R (5′-CTGAAGGCGGTCCTGCGATGACCGAACACCCCGCACA-3′). After first PCR amplification, gel purification was performed to recover target fragments. Due to the overlap between complementary sequences of primers L1307F and L1306R (marked with underline), second PCR amplification was carried out using recovered fragments as templates and primers L1306F and L1307R to obtain the fusion fragment LF-nocB. To generate a fusion fragment LF-nocB-RF, the right homologous fragment (RF) of nocB was amplified from genomic DNA of S. actuosus ATCC 25421 by PCR using primers 1308F (5′-GGCGTCGGCGCGGGTCATGGGGGCCTCCACGCTCAGATCCGCACC-3′) and 1308R (5′-GGATCCCGCCGCGACTTCACCTCGTCGTTCGTCGAGTTCGGCCAGGCCCTC-3′). Similarly, fusion between LF-nocB and RF was accomplished using primer pairs L1306F/L1308R to obtain fragment LF-nocB-RF, which was then cloned to yield pMD-1307 by T4 DNA ligase at 16 °C for 12 hours. After DNA sequencing, correct fragments digested by Hind III/BamH I were recovered and ligated into pKC1139 o generate pKL1307 according to the operational protocols. Then, the recombinant plasmid was introduced into S. actuosus L1120 by E. coli–Streptomyces conjugation. After double-crossover homologous recombination, the genotypes of the recombinant strain S. actuosus L1307 was further confirmed by PCR using primer pairs of nosAB-S2F (5′-GCGGTCGGGTGTGTCGAATCGTGCGGACTC-3′)/nosAB-S2R (5′-CCAACCGGGACGAGAAGGTCTTCGGCGAGG-3′) and DNA sequencing.Fermentation and analysis of metabolite production
Fermentation of WT and recombinant strains was carried out according to the method described previously.7,9 The products of WT and L1307 mutant were analyzed, quantified and verified according to the previous procedures.7,9Transcriptional analysis by RT-qPCR
Total RNA of WT and recombinant strains was extracted from mycelia in the fermentation medium after 48 h culturing. To obtain cDNA, DNase I treatment and reverse transcription were performed using PrimeScript™ RT reagent kit. The transcription levels of noc(s)B and nosA genes were assayed on a ABI StepOne Plus™ instrument (USA). RT-qPCR amplification was performed with 25 μL mixture containing 1 μg mL−1 template cDNA, 12.5 μL 2 × SYBR premix and 0.2 μM primers. The PCR program was as follows: 95 °C for 30 s, followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. Using 16s rRNA gene (GenBank no. KF924734) as an internal control, a 122 bp fragment was amplified using primers qPCRf1n (5′-ATACCGTGAGGTGGAGCGAAT-3′) and qPCRr1n (5′-AACGTATTCACCGCAGCAAT-3′). To assess the transcription level of nocB, a 138 bp internal fragment was amplified using primers of nocB-qF (5′-ACGTCCGCGCTGGTGTAGGTCATCCC-3′) and nocB-qR (5′-GGCCCTGTTCCAGCACCCGGACCAGC-3′). To measure the transcription level of nosB, a 138 bp internal fragment was amplified using primers of nosB-qF (5′-ACCTCCTCGGTGGTGACGGTCGTCCC-3′) and nosB-qR (5′-CGCCCTCTTCCAGCACCCCGACCAGC-3′), and to determine the transcription level of nosA, a 138 bp internal fragment was amplified using primers of nosA-qF (5′-ACGAACTCCTGCGGGCGGGTCAG-3′) and nosA-qR (5′-CCCGACCGAGCGCCGCTACTACA-3′). Relative transcription levels were calculated according to 2−ΔΔCt method.20Construction of fusion proteins
To detect the expression of NosB in S. actuosus ATCC 25421, a 2618 bp gene fragment containing full length of nosC gene and nosB gene (lacking TGA at C-terminus) was amplified from genomic DNA of S. actuosus ATCC 25421 by PCR. Then, recovered fragment was digested by Nde I/EcoR I and ligated into the same sites of pET28a(+) vector to generate plasmid pET28a-WT-cb. Green fluorescent protein gene fragment (gfp) was digested by EcoR I/Hind III from the plasmid pETDuet-gfp and ligated into the same sites of pET28a-WT-cb to generate target pET28a-WT-c-nosb-gfp. With the same procedure and restriction sites, recombinant plasmid pET28a-L1307-c-nocb-gfp was also constructed to detect the expression of NocB in S. actuosus L1307.To detect the expression of NosA in S. actuosus ATCC 25421 and L1307 mutant, 3070 bp and 2911 bp gene fragments amplified from genomic DNA of S. actuosus ATCC 25421 and S. actuosus L1307 using primer pairs of wtCB-28aF/wtCBA-28aR (5′-GAATTCCGCCGGCGGCCGGGAGGGGA-3′) were cloned into pMD-19T. Then, the target fragments were ligated into the pET28a(+) digested by Nde I/EcoR I to generate recombinant plasmids pET28a-WT-c-nosb-a and pET28a-L1307-c-nocb-a. Subsequently, the gfp fragment was ligated into both plasmids to generate recombinant pET28a-WT-c-nosb-a-gfp and pET28a-L1307-c-nocb-a-gfp, respectively.
Detection of fluorescence intensity
Four recombinant plasmids, pET28a-WT-c-nosb-gfp, pET28a-L1307-c-nocb-gfp, pET28a-WT-c-nosb-a-gfp and pET28a-L1307-c-nocb-a-gfp were transformed into E. coli BL21 (DE3) for protein expression. The expression was conducted at 20 °C, 220 rpm and 1 mM IPTG for 16 hours. Then, the resulting cells were harvested and disrupted by cell disrupter. After centrifugation, the resulting supernatants were collected and 200 μL of each sample was used for the determination of fluorescence intensity with excitation wavelength of 397 nm and emission wavelength of 506 nm.21 The fluorescence intensity was recorded by subtracting the background from control.Expression and purification of NocB and NosB
pET28a vector was selected to express NocB and NosB with a His6-tagged sequence at the N-terminus of both enzymes. The pET28a and nosB (or nocB) gene from genomic DNA of S. actuosus ATCC 25421 (or genomic DNA of Nocardia sp. ATCC 202099) were digested by Nde I and Hind III, and ligated with T4DNA ligase. The constructed pET28a-nosB (or pET28a-nocB) was transformed into E. coli BL21 (DE3) for expression, which was conducted at 16 °C, 220 rpm and 0.5 mM IPTG for 16 hours. The resulting cells expressing NosB (or NocB) were harvested by centrifugation (12000 × g) for 10 min at 4 °C.
E. coli cells (1.0 g) expressing NosB were suspended in 10 mL of 0.05 M potassium phosphate buffer (pH 7.4). The cells were disrupted by passing through a high pressure cell press at 30 kpsi. Cell-free extract (1 mL) was obtained by centrifugation at 13500 × g for 30 min and applied onto a Ni-Sepharose 6 FF column (1 mL). After 4 h absorption at 4 °C, the column was eluted with a 10 mL linear gradient of (0–0.1 M) imidazole in 50 mM potassium phosphate buffer (pH 7.4). Fractions containing NosB were pooled and concentrated to final volume of 1 mL, which was loaded to HiTrap™ Q FF column (1.0 mL) and eluted with 10 mL of a linear gradient of 0–1 M NaCl in 50 mM potassium phosphate buffer (pH 7.4). The fractions containing NosB were collected and concentrated. The purity of NosB was judged by SDS-PAGE. The purification of NocB was conducted with the same procedure as NosB, except that the linear gradient of imidazole for Ni-Sepharose column was 0–0.12 M in 0.05 M potassium phosphate buffer (pH 7.4). Subsequently, enzymatic reaction catalyzed by NocB using intermediate 3 as a substrate was performed according to previous report.9
Kinetic studies for NosB and NocB
For all assays, enzyme activity was defined as one unit representing the production of one μmole of nosiheptide per min per milligram protein. The nosiheptide product catalyzed by NosB or NocB was analyzed and quantified by HPLC.9 To determine the kinetic constants for NosB, reaction mixtures contained 15 mM D-glucose-6-phosphate, 1 U glucose-6-phosphate dehydrogenase, 0.3 U ferredoxin–NADP reductase, 60 μg ferredoxin, 3.0 M NADPH, various concentrations of intermediate 3 (41.5 μM to 165.8 μM), 410 μg NosB and 50 mM potassium phosphate buffer (pH 7.4) in a final volume of 1.0 mL. To determine the kinetic constants for NocB, the same reaction system were constructed with 1.45 mg mL−1 NocB. All reactions were performed at 220 rpm and 30 °C for 20 min, and terminated by addition of 1 mL ethanol for HPLC analysis.Acknowledgements
This work was supported by the National Key Project on Science and Technology of China (2012ZX09103101-030), the National Science Foundation of China (81502961), the PAPD of Jiangsu Province and the Project of University Collaborative Innovation Center of Jiangsu Province (Biological Medicine Center).References
- M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685–714 CrossRef CAS PubMed.
- P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- F. Benazet, M. Cartier, J. Florent, C. Godard, G. Jung, J. Lunel, D. Mancy, C. Pascal, J. Renaut and P. Tarridec, Experientia, 1980, 36, 414–416 CrossRef CAS PubMed.
- T. Sasaki, T. Otani, H. Matsumoto, N. Unemi, M. Hamada, T. Takeuchi and M. Hori, J. Antibiot., 1998, 51, 715–721 CrossRef CAS PubMed.
- Y. Yu, L. Duan, Q. Zhang, R. J. Liao, Y. Ding, H. X. Pan, E. Wendt-Pienkowski, G. L. Tang, B. Shen and W. Liu, ACS Chem. Biol., 2009, 4, 855–864 CrossRef CAS PubMed.
- Y. Ding, Y. Yu, H. Pan, H. X. Guo, Y. M. Li and W. Liu, Mol. BioSyst., 2010, 6, 1180–1185 RSC.
- W. Y. Liu, M. Ma, Y. J. Xue, N. Liu, S. Z. Wang and Y. J. Chen, ChemBioChem, 2013, 14, 573–576 CrossRef CAS PubMed.
- Y. Yu, H. Guo, Q. Zhang, L. Duan, Y. Ding, R. Liao, C. Lei, B. Shen and W. Liu, J. Am. Chem. Soc., 2010, 132, 16324–16326 CrossRef CAS PubMed.
- W. Y. Liu, Y. J. Xue, M. Ma, S. Z. Wang, N. Liu and Y. J. Chen, ChemBioChem, 2013, 14, 1544–1547 CrossRef CAS PubMed.
- D. R. Nelson, L. Koymans, T. Kamataki, J. J. Stegeman, R. Feyereisen, D. J. Waxman, M. R. Waterman, O. Gotoh, M. J. Coon, R. Estabrook, I. C. Gunsalus and D. W. Nebert, Pharmacogenetics, 1996, 6, 1–42 CrossRef CAS PubMed.
- D. R. Nelson, Cytochrome P450 nomenclature, Methods Mol. Biol., 2004, 320, 1–10 Search PubMed.
- M. C. Wei, J. Deng, S. Z. Wang, N. Liu and Y. J. Chen, Biotechnol. Lett., 2011, 33, 585–591 CrossRef CAS PubMed.
- Q. Zhang and W. Liu, Nat. Prod. Rep., 2013, 30, 218–226 RSC.
- C. Li and W. L. Kelly, Nat. Prod. Rep., 2010, 27, 153–164 RSC.
- H. Wu, S. Qu, C. Y. Lu, H. J. Zheng, X. F. Zhou, L. Q. Bai and Z. X. Deng, BMC Genomics, 2012, 13, 337–351 CrossRef CAS PubMed.
- P. Claudine, D. Arnaud, L. Jean and P. Thierry, J. Am. Chem. Soc., 1977, 99, 6418–6423 CrossRef.
- O. D. Hensens and G. Albers-Schönberg, Tetrahedron Lett., 1978, 19, 3649–3652 CrossRef.
- J. E. Leet, W. Li, H. A. Ax, J. A. Matson, S. Huang, R. Huang, J. L. Cantone, D. Drexler, R. A. Dalterio and K. S. Lam, J. Antibiot., 2003, 56, 232–242 CrossRef CAS PubMed.
- T. Kieser, M. J. Bibb, M. J. Buttner, K. F. Chater and D. A. Hopwood, Int. Microbiol., 2000, 3, 260–261 Search PubMed.
- K. J. Livaka and T. D. Schmittgenb, Methods, 2001, 25, 402–408 CrossRef PubMed.
- G. H. Patterson, S. M. Knobel, W. D. Sharif, D. S. Kain and D. W. Piston, Biophys. J., 1997, 73, 2782–2790 CrossRef CAS PubMed.
Footnote |
| † Xuri Wu and Peiyu Huang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |