
Investigation into the mechanism of polyoxotungstates-catalyzed cyclooctene epoxidation by ESI-MS†
LinYuan Fan,
YaYu Hong,
Jie Cao* and
ChangWen Hu*
Key Laboratory of Cluster Science, Ministry of Education of China, Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, School of Chemistry, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: jcao@bit.edu.cn; cwhu@bit.edu.cn; Fax: +86-10-81381380; Tel: +86-10-68912631
First published on 6th June 2016
Abstract
The reaction process of cyclooctene epoxidation was real-time monitored by ESI-MS using H3PW12O40 and H2WO4 as catalysts. By observing the species changes of the catalysts in the reaction solutions and the combination of catalysts with the substrate, a catalytic reaction mechanism of polyoxometalate clusters was proposed from the aspect of aggregation and dissociation. When H3PW12O40 was used as the catalyst, it was degraded into minor peroxo-species [PW4O16−x(O2)x]3− (x = 0, 2, 4, 6, 8) which performed as the active center of the epoxidation of cyclooctene. When H2WO4 was used as the catalyst, it reacted with H2O2 and generated mononuclear tungsten peroxo-species [HWO2(O2)2]− and [HWO2(O2)3]−. After the addition of cyclooctene, the peroxo-species catalyzed the epoxidation of cyclooctene, generating 1,2-epoxycyclooctane and combining with 1,2-epoxycyclooctane to form the intermediate [HW2(O2)2O5(C8H14O)2]−.
Introduction
Polyoxometalates (POMs) consist of metal oxide clusters based on the early transition metals Mo, W, V, Nb and Ta, and have provoked significant interest in catalysis due to their redox and acid–base properties.1,2 They have been used as homogeneous catalysts for a wide variety of organic substrate oxidations, such as alkane, olefins and alcohols etc.3–5 Epoxides are products of olefin epoxidation, and are valuable compounds used as resins, paints, surfactants, and intermediates in various organic syntheses.6 In contrast to synthetic methods with low atom efficiencies that use stoichiometric reagents, catalytic epoxidation of alkenes with hydrogen peroxide (H2O2) has attracted much attention, because H2O2 has a high content of active oxygen species and only water is formed as a by-product. Therefore, a large number of H2O2-based epoxidation systems that use POM catalysts have been developed, such as H3PW12O40 and TBA4H4[γ-SiW10O36] etc.7–13Tungsten-based epoxidation systems with hydrogen peroxide have attracted much attention because of their high reactivity for olefins and inherent poor activity for the decomposition of hydrogen peroxide.12 In the catalytic process, polyoxotungstates act as catalyst precursors, which decompose into the monomeric, dimeric and tetrameric peroxo species by reaction with hydrogen peroxide.14 The [PO4{WO(O2)2}4]3−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7,15,16 and [W2O3(O2)4(H2O)2]2−17,18 peroxotungstates, which can catalyze the epoxidation, have been isolated and characterized crystallographically. Although the peroxo-species in the TBA2[W2O3(O2)4(H2O)2]/H3[PW12O40]-based epoxidation systems with H2O2 have been investigated by 31P/17O NMR and Density Functional Theory (DFT) calculations,19–21 the intermolecular oxidation reaction providing further proof that peroxo-species are reactive species has not yet been probed.
7,15,16 and [W2O3(O2)4(H2O)2]2−17,18 peroxotungstates, which can catalyze the epoxidation, have been isolated and characterized crystallographically. Although the peroxo-species in the TBA2[W2O3(O2)4(H2O)2]/H3[PW12O40]-based epoxidation systems with H2O2 have been investigated by 31P/17O NMR and Density Functional Theory (DFT) calculations,19–21 the intermolecular oxidation reaction providing further proof that peroxo-species are reactive species has not yet been probed.
Mass spectrometry, as a new characterization method for the observation of active species,22,23 has been previously applied to the study of catalytic mechanisms. For example, Cronin observed the generation of an intermediate Fe(v)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species within a synthetic non-haem complex and its reaction with an olefin by variable temperature mass spectrometry.24 In this paper, we investigate the reaction mechanism for the epoxidation of cyclooctene with H2O2 catalyzed by H3PW12O40 and H2WO4 with mass spectroscopy for the aspects of decomposition and aggregation and explored the effects of reaction time, ratio of catalyst in the catalytic system.
O species within a synthetic non-haem complex and its reaction with an olefin by variable temperature mass spectrometry.24 In this paper, we investigate the reaction mechanism for the epoxidation of cyclooctene with H2O2 catalyzed by H3PW12O40 and H2WO4 with mass spectroscopy for the aspects of decomposition and aggregation and explored the effects of reaction time, ratio of catalyst in the catalytic system.
Results and discussion
Epoxidation of cyclooctene by H2O2 catalyzed by polyoxometalates
The oxidation of cyclooctene by H2O2 was examined in the presence of different amounts of H3PW12O40 (catalyst 1) and H2WO4 (catalyst 2). The results of these reactions are summarized in Table 1 and Fig. S1 and S2.† The reaction cannot proceed without a catalyst (Entry 5). These two kinds of polyoxometalates were successful catalysts evaluated in terms of both conversion of cyclooctene and yield of 1,2-epoxycyclooctane when the amount of catalyst is low (Entry 1 and 3). However, increasing the amount of catalyst led to a significant decrease of 1,2-epoxycyclooctane yield (Entry 2 and 4) without any other by-products increased (Fig. S1 and S2†). This phenomenon of non-conservation in GC analysis strongly suggests that the catalyst and 1,2-epoxycyclooctane combined to form new complexes, which may be difficult to gasify in GC.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Concentration [mM] | Conv.b [% mol] | Yieldb [% mol] | |
| [POM] | [W] | ||||
| a Reaction condition: refer to the Experimental section.b Determined by GC with area normalization. | |||||
| 1 | H3PW12O40 | 0.005 | 0.06 | 86.71 | 76.30 |
| 2 | H3PW12O40 | 0.1 | 1.2 | 84.63 | 20.41 |
| 3 | H2WO4 | 0.06 | 0.06 | 80.52 | 71.21 |
| 4 | H2WO4 | 1.2 | 1.2 | 80.37 | 35.29 |
| 5 | None | 0 | 0 | 0 | 0 |
MS spectra of the catalytic solution
To confirm our speculation, the final catalytic solution was characterized by electrospray ionization mass spectrometry (ESI-MS). In the ESI-MS spectra of the catalytic solution catalyzed by H3PW12O40 (Fig. 1A) and H2WO4 (Fig. 1B), the most intense peak (m/z 765.1) with an isotopic distribution (Fig. S3A†) that agrees with the pattern calculated for [HW2(O2)2O5(C8H14O)2]− can be seen as a combination of peroxo-species {W2} and 1,2-epoxycyclooctane (Fig. S3B†). Besides, [PW12O40]3− (m/z 958.7) and [HPW12O40]2− (m/z 1438.6) also exist in the catalytic system 1 (Fig. 1A), while species belonging to [WO4]2− cannot be measured in the system 2 (Fig. 1B) for the low solubility of H2WO4. In order to infer the formation of [HW2(O2)2O5(C8H14O)2]−, we replaced cyclooctene with 1,2-epoxycyclooctane in the catalytic solution, i.e. 1,2-epoxycyclooctane, H2O2 and catalysts dissolved in CH3CN. The MS spectra of the final solution were the same as the system with cyclooctene as the substrate (Fig. S4†) and no other organic product was monitored in the GC spectra, which tends to indicate that [HW2(O2)2O5(C8H14O)2]− was assigned to a combination of peroxo-species with a product of 1,2-epoxycyclooctane.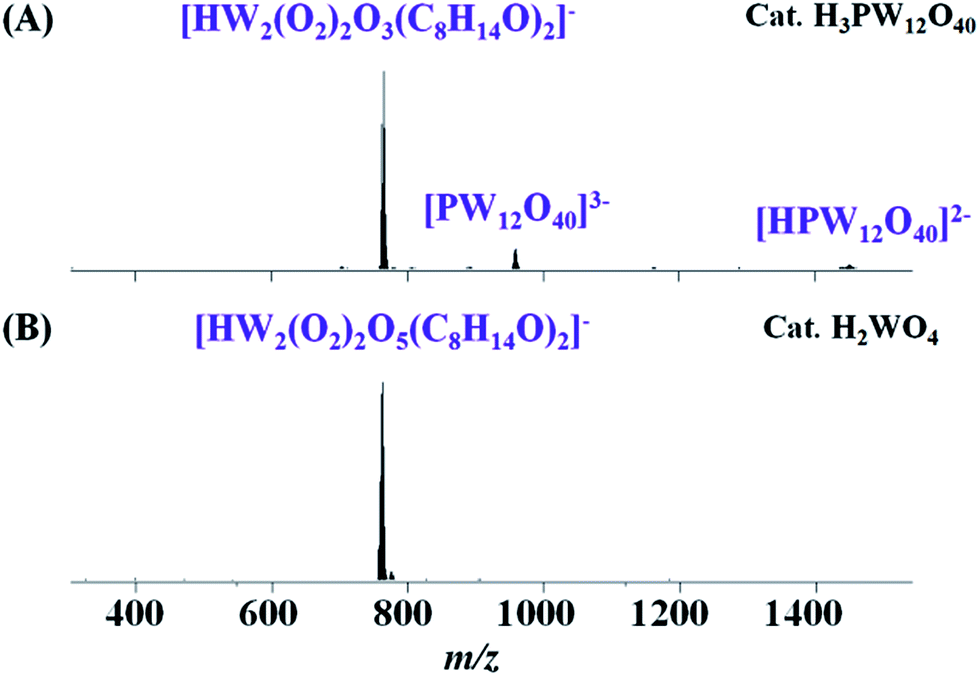 | ||
| Fig. 1 MS spectra of catalytic solution epoxidation reaction of cyclooctene after the epoxidation reaction. Reaction conditions: cyclooctene (1 mmol), H2O2 (1 mmol), CH3CN solvent (2 mL), 50 °C, 4 h. (A) H3PW12O40 (0.005 mmol) as catalyst; (B) H2WO4 (0.06 mmol) as catalyst. | ||
To further probe the formula of the [HW2(O2)2O5(C8H14O)2]− anions generated from the different catalysts, CID experiments on [HW2(O2)2O5(C8H14O)2]− were performed (Fig. 2). The CID spectra of [HW2(O2)2O5(C8H14O)2]− with H3PW12O40 as the catalyst (Fig. 2A) and H2WO4 as the catalyst (Fig. 2B) were similar, proving that [HW2(O2)2O5(C8H14O)2]− anions have the same structure in different catalytic systems. Besides, the CID spectra of the [HW2(O2)2O5(C8H14O)2]− from cyclooctene and 1,2-epoxycyclooctane with H3PW12O40 (Fig. S5A and B†) and H2WO4 (Fig. S5C and D) as the catalyst are almost the same.
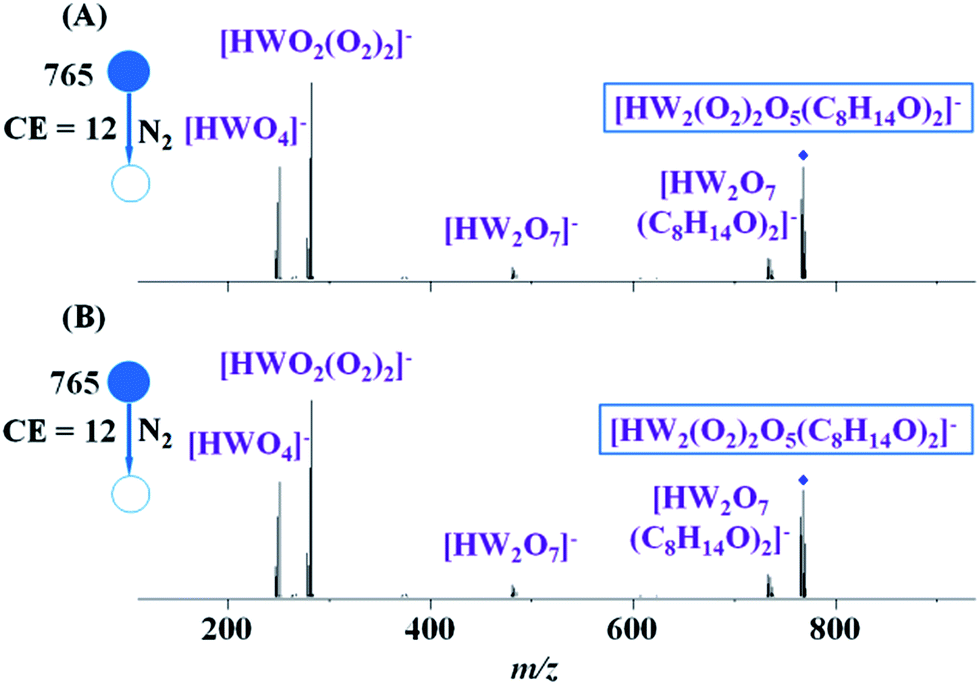 | ||
| Fig. 2 CID mass spectra of [HW2(O2)2O5(C8H14O)2]− generated from different reaction solutions. (A) H3PW12O40 (0.005 mmol) as the catalyst; (B) H2WO4 (0.06 mmol) as the catalyst. Collision energy = 12 eV, isolation width = 9. The parent ion (denoted by a diamond) is shown in a blue square box in each spectrum. | ||
Real-time monitoring on the process of cyclooctene epoxidation
The reaction solutions of 1 and 2 (H3PW12O40 and H2WO4 as catalyst respectively) were real-time monitored by ESI-MS to reveal the change of species during the reaction, identifying the species requisite for cyclooctene epoxidation and the complex species that catalytic active species combined 1,2-epoxycyclooctane.Firstly, the reaction solutions were real-time monitored using ESI-MS by one-step (Fig. S6 and S7†). First, the catalyst, H2O2 (1 mmol) and cyclooctene (1 mmol) were dissolved in CH3CN. Then the samples were taken from the solution at intervals. The real-time monitoring of the solution of catalyst 1 (Fig. S6†) showed that the peaks assigned to [HW2(O2)2O5(C8H14O)2]− appeared at the 5th min and its abundance increased progressively. At last, it became the most intense peak at about the 60th min. The corresponding GC spectra showed that cyclooctene was completely converted to 1,2-epoxycyclooctane at about the 60th min (Fig. S9A†). The ESI-MS spectra of the real-time monitoring of the solution containing catalyst 2 (Fig. S7†) were similar to that with catalyst 1. The peaks assigned to [HW2(O2)2O5(C8H14O)2]− appeared at the 5th min and became the most intense peaks for the low solubility of H2WO4. The corresponding GC spectra showed that cyclooctene was converted to 1,2-epoxycyclooctane completely at about the 3rd hour (Fig. S10A†).
To further investigate how the [HW2(O2)2O5(C8H14O)2]− formed, the catalytic reactions were real-time monitored using ESI-MS by two-step. H2O2 and the catalyst were mixed previously and reacted for 4 h, and cyclooctene was added to the solution.
For the two-step catalytic system 1 (H3PW12O40 as catalyst), the ESI-MS monitoring data (Fig. 3) was illustrated as follows. Firstly, when H3PW12O40 (0.005 mmol) was dissolved in CH3CN, all major peaks in the mass spectrum can be assigned to [PW12O40]3− (m/z 958.7) and [HPW12O40]2− (m/z 1438.6), which means the {PW12} unit is stable in CH3CN at this step (Fig. 3A). After adding 1 mmol H2O2 to the previous solution with stirring at 50 °C for 5 min, some new species formed during this process. As the spectrum (Fig. 3B) shows, the new peaks at m/z 340.9–383.6 can be assigned to [PW4O16−x(O2)x]3− (x = 0, 2, 4, 6, 8), with m/z 340.9 for [PW4O16]3−, m/z 351.6 for [PW4O14(O2)2]3−, m/z 362.2 for [PW4O12(O2)4]3−, m/z 372.9 for [PW4O10(O2)6]3−, m/z 383.6 for [PW4O8(O2)8]3−, meaning that the parent {PW12} can react with H2O2, yielding the degraded peroxo-species [PW4O16−x(O2)x]3−. Upon continuous heating of the solution for 4 h, the intensity of the peaks assigned to [PW4O16−x(O2)x]3− (x = 0, 2, 4, 6, 8) gradually increased and became the main peaks (Fig. 3C). After adding cyclooctene (1 mmol) to the former reaction solution of {PW12} + H2O2 with stirring at 50 °C for 5 min, the new species related to [HW2(O2)2O5(C8H14O)2]− (m/z 765.1) emerged accompanied with a decline of [PW4O16−x(O2)x]3− (Fig. 3D). After continuous heating of the solution for 4 h, the intensity of [HW2(O2)2O5(C8H14O)2]− increased (Fig. 3E), indicating that the [HW2(O2)2O5(C8H14O)2]− intermediate was a combination of peroxo-species {W2} and product 1,2-epoxycyclooctane. As 1,2-epoxycyclooctane increased in the system, the amount of [HW2(O2)2O5(C8H14O)2]− gradually increased. Besides, the corresponding GC spectra showed that cyclooctene quickly completed the conversion to 1,2-epoxycyclooctane at about the 10th min (Fig. S9B†), which supported that [PW4O16−x(O2)x]3− (x = 0, 2, 4, 6, 8) were active species in the catalytic system.
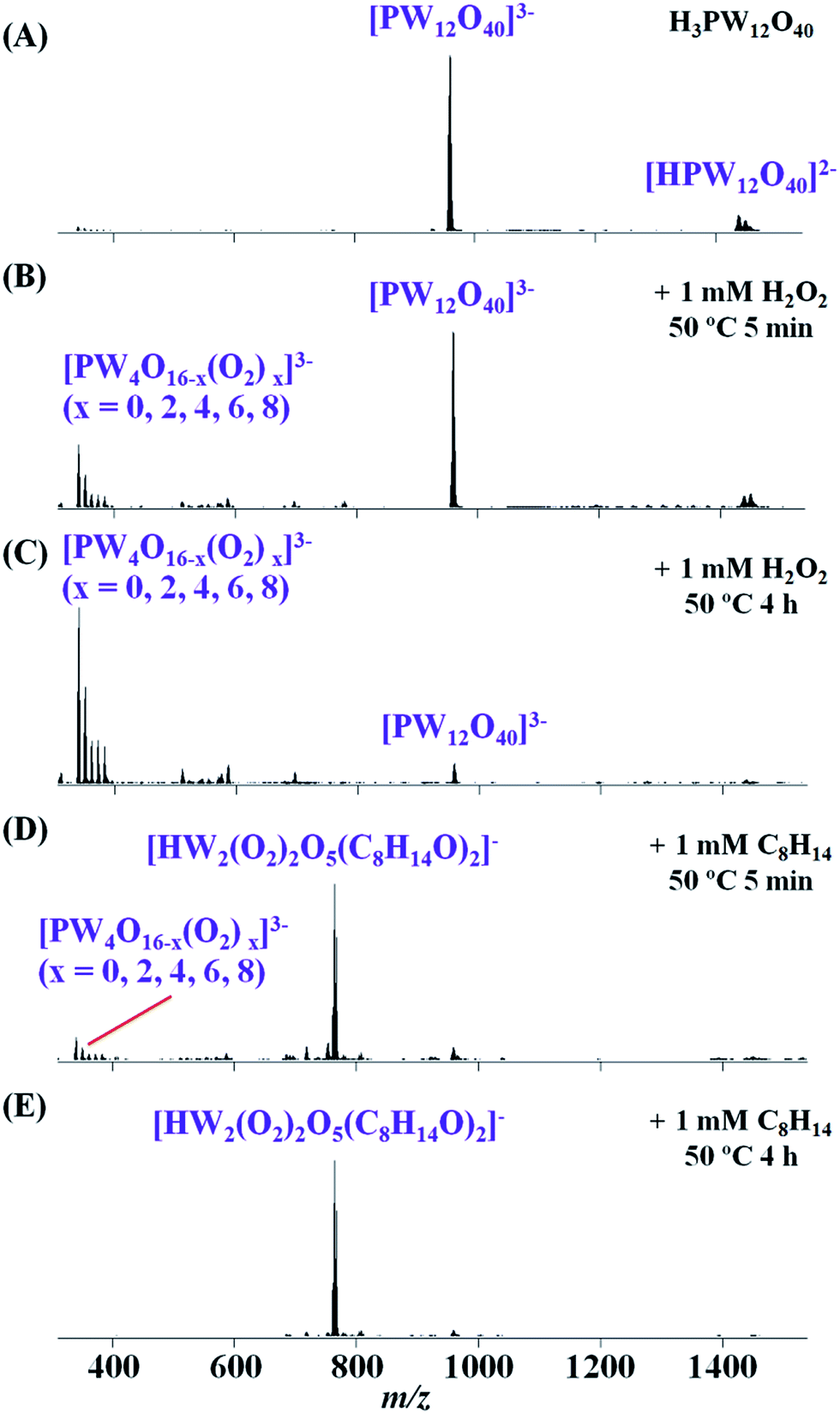 | ||
| Fig. 3 Real-time monitoring on the cyclooctene epoxidation by two-step of system 1. (A) H3PW12O40 (0.005 mmol) dissolved in CH3CN; (B) stirring 5 min at 50 °C after the addition of H2O2 (1 mmol); (C) stirring 4 h; (D) stirring further 5 min after the addition of cyclooctene (1 mmol); (E) stirring further 4 h. | ||
For the catalytic system 2 (H2WO4 as catalyst), the real-time ESI-MS monitoring data (Fig. 4) by two-step was similar to system 1. Firstly, when the catalyst H2WO4 (0.06 mmol) mixed with CH3CN, there are only weak peaks assigned to [HWO4]− as a result of the low solubility of H2WO4 in CH3CN (Fig. 4A). After adding 1 mmol H2O2 to the H2WO4 solution with stirring at 50 °C for 5 min, a remarkable change occurred in the mixed solution. As the spectrum (Fig. 4B) shows, the new peaks at m/z 280.9–296.9 can be assigned to [HWO2(O2)2]− (m/z 280.9) and [HWO(O2)3]− (m/z 296.9). This result indicates that H2WO4 can react with H2O2, producing soluble peroxo-species [HWO4−x(O2)x]− (x = 2, 3). Besides, the reaction was performed under heating, which also increases the solubility of H2WO4. Upon continuous heating of the solution for 4 h, the intensity of the peaks assigned to [HWO(O2)3]− gradually increased and became the main peak (Fig. 4C). Then, after adding cyclooctene (1 mmol) to the mixed reaction solution of H2WO4 + H2O2 with stirring at 50 °C for 5 min, a new species belonging to [HW2(O2)2O5(C8H14O)2]− (m/z 765.1) emerged accompanied with a decrease of [HWO4−x(O2)x]− (Fig. 4D). After continuous heating of the solution for 4 h, the intensity of [HW2(O2)2O5(C8H14O)2]− increased (Fig. 4E). The corresponding GC spectra showed that cyclooctene converted to 1,2-epoxycyclooctane completely at about the 30th min (Fig. S10B†), which supported the speculation that [HWO4−x(O2)x]− (x = 2, 3) were active species in the catalytic system.
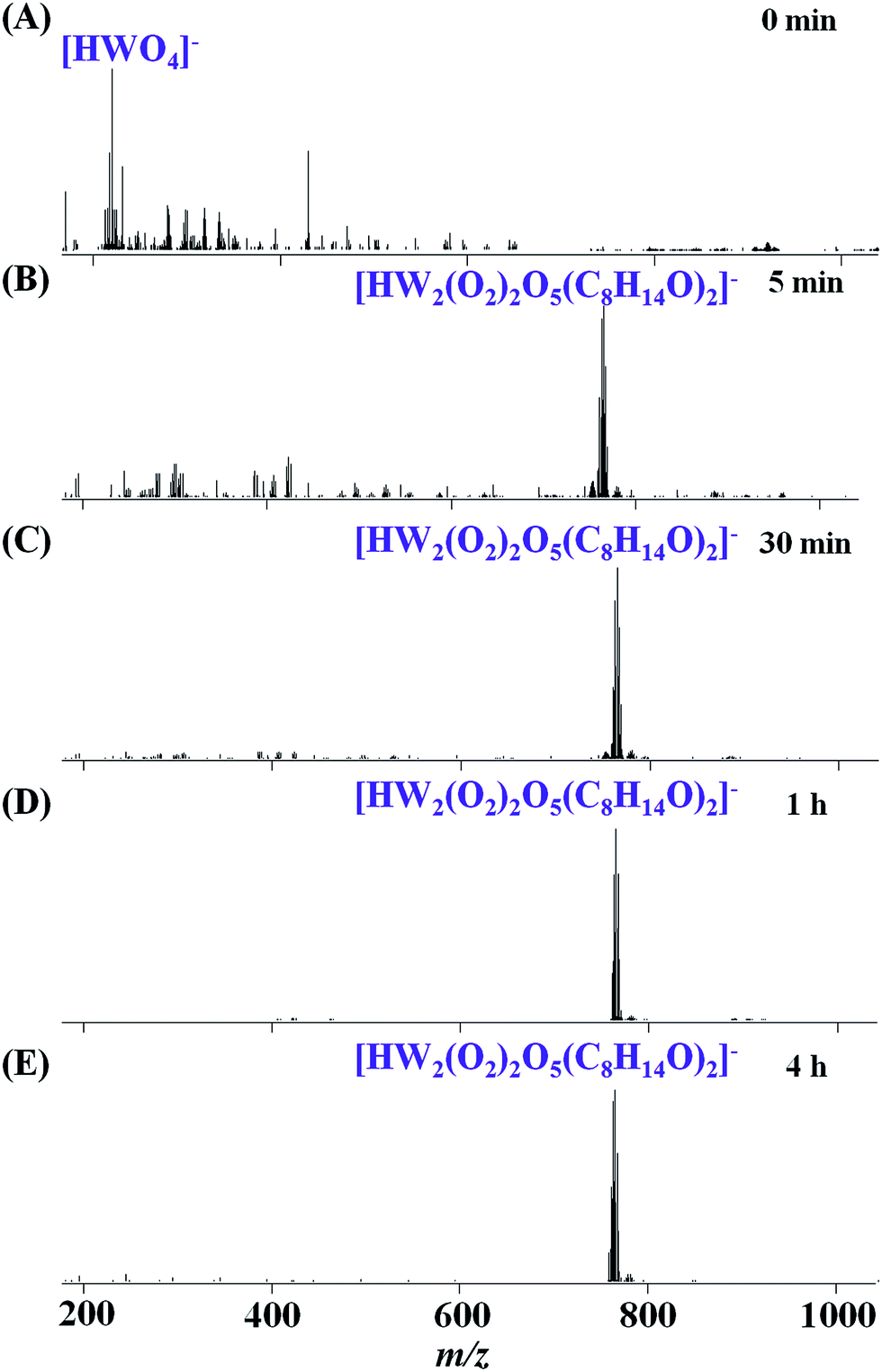 | ||
| Fig. 4 Real-time monitoring of the cyclooctene epoxidation by a two-step of system 2. (A) H2WO4 (0.06 mmol) mixed with CH3CN; (B) stirring for 5 min at 50 °C after the addition of H2O2 (1 mmol); (C) stirring for 4 h; (D) stirring for a further 5 min after the addition of cyclooctene (1 mmol); (E) stirring for a further 4 h. | ||
In summary, using Keggin type heterpolyoxotungstates H3PW12O40 and mononuclear tungstate H2WO4 as catalysts, the reaction process of cyclooctene epoxidation was real-time monitored by ESI-MS (Scheme 1). When H3PW12O40 was used as catalyst, it was degraded into minor peroxo-species [PW4O16−x(O2)x]3− (x = 0, 2, 4, 6, 8) which performed as the active center of the epoxidation of cyclooctene. After the addition of cyclooctene, the peroxo-species and 1,2-epoxycyclooctane formed intermediate [HW2(O2)2O5(C8H14O)2]−. When H2WO4 was used as the catalyst, it reacted with H2O2 and generated mononuclear tungsten peroxo-species [HWO2(O2)2]− and [HWO2(O2)3]−, which catalyzed the epoxidation of cyclooctene generating 1,2-epoxycyclooctane. After the addition of cyclooctene, the peroxo-species and 1,2-epoxycyclooctane formed intermediate [HW2(O2)2O5(C8H14O)2]−. In the presence of a large amount of catalyst, the non-conservation of olefin-epoxide in GC analysis also confirmed the existence of a stable intermediate. Future work will be focused on the structure and effect of the intermediate [HW2(O2)2O5(C8H14O)2]− in the catalytic system.
 | ||
| Scheme 1 Speculated mechanism of cyclooctene epoxidation using H3PW12O40 and H2WO4 as catalysts. | ||
Experimental
Materials
All chemicals were purchased from commercial sources and used without further purification.Catalytic oxidation of cyclooctene
Oxidations of cyclooctene were carried out in a thermostated sealed glass tube equipped with a magnetic stirrer. The initial ratio of cyclooctene (1 mmol) oxidation by H2O2 (1 mmol, added as 30 wt% aqueous solution) at 50 °C for 4 h in 2 mL CH3CN was used to assess the relative catalytic activities of the different POMs. Products were identified from their mass (GC-MS) spectra. The yields of the product 1,2-epoxycyclooctane as well as the conversion of the initial substrate cyclooctene were quantified by GC with area normalization.Mass spectrometry
All MS data were obtained in the negative ion mode using an Agilent 6520 Q-TOF LC/MS. An electrospray source was used to collect data under the conditions specified below. The detector was a time-of-flight and all data were processed using the Agilent qualitative analysis B.04.00 software, while simulated isotope patterns were investigated using Agilent Isotope Pattern software and Molecular Weight Calculator. The following parameters were consistent for all ESI-MS scans given below. The calibration solution used was Agilent ESI tuning mix solution, enabling calibration between ∼100 m/z and 3000 m/z. This solution was diluted 100:1 with acetonitrile. Samples were introduced into the MS via an automatic sampler. The electrospray settings were set with the drying nitrogen gas temperature at 300 °C. The ion polarity for all MS scans recorded was negative, with the voltage of the capillary tip set at 3500 V, skimmer voltage at 65 V, RF at 750 V, fragmentor at 80 V. All theoretical peak assignments were determined via comparison of the experimentally determined isotopic patterns for each peak, with simulated isotopic patterns. CID experiments were performed using N2 as the target gas. Collisional energy voltage was 12 V for [HW2(O2)2O5(C8H14O)2]− precursors.
Gas chromatography
GC analyses were performed on Shimadzu GC-2014C with a FID detector equipped with an Rtx-1701 Sil capillary column.Acknowledgements
The authors thank the National Natural Science Foundation of China (21173021, 21231002, 21276026, 21371025), 973 Program (2014CB932103), the 111 Project (B07012) and the Fundamental Research Grant (20121942006) by Beijing Institute of Technology.Notes and references
- M. T. Pope, Heteropoly and Isopoly Oxometalates, Springer-Verlag, Berlin, 1983 Search PubMed.
- C. L. Hill, Chem. Rev., 1998, 98, 1–390 CrossRef CAS PubMed.
- K. Kamata, K. Yonehara, Y. Sumida, K. Yamaguchi, S. Hikichi and N. Mizuno, Science, 2003, 300, 964–970 CrossRef CAS PubMed.
- R. Ben-Daniel, A. M. Khenkin and R. Neumann, Chem.–Eur. J., 2000, 6, 3722–3728 CrossRef CAS.
- K. Yamaguchi, S. Shinachi and N. Mizuno, Chem. Commun., 2004, 424–425 RSC.
- J. E. Bäckvall, Modern Oxidation Methods, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed.
- R. B. Brown and C. L. Hill, J. Org. Chem., 1988, 53, 5762–5768 CrossRef CAS.
- A. C. Dengel, W. P. Griffith and B. C. Parkin, J. Chem. Soc., Dalton Trans., 1993, 2683–2688 RSC.
- R. Ishimoto, K. Kamata and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 4662–4665 CrossRef CAS PubMed.
- N. Mizuno, C. Nozaki, I. Kiyoto and M. Misono, J. Catal., 1999, 182, 285–288 CrossRef CAS.
- R. Neumann and M. Gara, J. Am. Chem. Soc., 1995, 117, 5066–5074 CrossRef CAS.
- T. M. Anderson, X. Zhang, K. I. Hardcastle and C. L. Hill, Inorg. Chem., 2002, 41, 2477–2488 CrossRef CAS PubMed.
- D. C. Duncan, R. C. Chambers, E. Hecht and C. L. Hill, J. Am. Chem. Soc., 1995, 117, 681–691 CrossRef CAS.
- N. Mizuno, K. Yamaguchi and K. Kamata, Coord. Chem. Rev., 2005, 249, 1944–1956 CrossRef CAS.
- Y. Ding, W. Zhao, H. Hua and B. C. Ma, Green Chem., 2008, 10, 910–913 RSC.
- Y. Ding, B. C. Ma, D. J. Tong, H. Han and W. Zhao, Aust. J. Chem., 2009, 62, 739–746 CrossRef CAS.
- K. Kamata, K. Yamaguchi, S. Hikichi and N. Mizuno, Adv. Synth. Catal., 2003, 345, 1193–1196 CrossRef CAS.
- K. Kamata, K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2004, 10, 4728–4734 CrossRef CAS PubMed.
- C. Venturello and R. D’Alosio, J. Org. Chem., 1988, 53, 1553–1557 CrossRef CAS.
- Y. Ishii, K. Yamawaki, T. Ura, H. Yamada, T. Yoshida and M. Ogawa, J. Org. Chem., 1988, 53, 3587–3593 CrossRef CAS.
- R. Ishimoto, K. Kamata and N. Mizuno, Eur. J. Inorg. Chem., 2013, 10–11, 1943–1950 CrossRef.
- L. Y. Fan, Z. G. Lin, J. Cao and C. W. Hu, Inorg. Chem., 2016, 55(6), 2900–2908 CrossRef CAS PubMed.
- Q. D. Jia, J. Cao, Y. P. Duan and C. W. Hu, Dalton Trans., 2015, 44, 553–559 RSC.
- I. Prat, J. S. Mathieson, M. Güell, X. Ribas, J. M. Luis, L. Cronin and M. Costas, Nat. Chem., 2011, 3, 788–793 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional GC spectra of the epoxidation reaction of cyclooctene; comparison of calculated and observed mass spectra; CID mass spectra; real-time monitoring on the cyclooctene epoxidation by one-step; conversion-time curve of cyclooctene epoxidation. See DOI: 10.1039/c6ra09561e |
| This journal is © The Royal Society of Chemistry 2016 |