
Hydrogen-terminated mesoporous silicon monoliths with huge surface area as alternative Si-based visible light-active photocatalysts†
Abstract
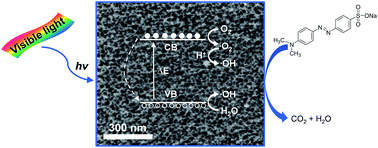
Silicon-based nanostructures and their related composites have drawn tremendous research interest in solar energy storage and conversion. Mesoporous silicon with a huge surface area of 400–900 m2 g−1 developed by electrochemical etching exhibits excellent photocatalytic ability and stability after 10 cycles in degrading methyl orange under visible light irradiation, owing to its unique mesoporous network, abundant surface hydrides and efficient light harvesting. This work showcases the profound effects of surface area, crystallinity, pore topology on charge migration/recombination and mass transportation. Therein the ordered 1D channel array has outperformed the interconnected 3D porous network by greatly accelerating the mass diffusion and enhancing the accessibility of the active sites on the extensive surfaces.
 Please wait while we load your content...
Please wait while we load your content...

