
Selective production of aromatic hydrocarbons from catalytic pyrolysis of biomass over Cu or Fe loaded mesoporous rod-like alumina
Surachai Karnjanakoma,
Asep Bayua,
Pairuzha Xiaoketia,
Xiaogang Haoc,
Suwadee Kongparakuld,
Chanatip Samartd,
Abuliti Abudulaa and
Guoqing Guan*ab
aGraduate School of Science and Technology, Hirosaki University, 1-Bunkyocho, Hirosaki 036-8560, Japan
bNorth Japan Research Institute for Sustainable Energy (NJRISE), Hirosaki University, 2-1-3, Matsubara, Aomori 030-0813, Japan. E-mail: guan@hirosaki-u.ac.jp; Fax: +81-17-735-5411; Tel: +81-17-762-7756
cDepartment of Chemical Engineering, Taiyuan University of Technology, Taiyuan 030024, China
dDepartment of Chemistry, Faculty of Science and Technology, Thammasat University, Pathumtani 12120, Thailand
First published on 13th May 2016
Abstract
The selective production of aromatic hydrocarbons from bio-oil derived from the fast pyrolysis of sunflower stalks over Cu or Fe-modified mesoporous rod-like alumina catalysts was investigated. Uniform mesoporous rod-like alumina with different pore sizes were successfully synthesized using a hydrothermal method with the assistance of Pluronic P123 surfactant. A high relative total hydrocarbon amount of about 59% in the upgraded bio-oil was obtained when pure mesoporous Al2O3 with a uniform pore size of 5.81 nm was used. Mesoporous Al2O3 with a larger pore size resulted in more polycyclic aromatic hydrocarbons (PAHs) such as indenes and naphthalenes being generated. Cu or Fe loaded Al2O3 with a loading amount in the range of 1–2.5 wt% showed a high selectivity towards monocyclic aromatic hydrocarbons (MAHs) such as benzene, toluene and xylenes (BTXs) over 80%. By using 2.5 wt% Cu/Al2O3-0.01, the highest relative total hydrocarbon amount reached 89%, which consisted of about 84% aromatic hydrocarbons and 4.9% aliphatic hydrocarbons. Both catalysts showed good catalytic stability and regeneration properties. A catalytic system with high effectiveness and long-term stability was expected to be obtained to convert the oxygenated compounds in bio-oil to high value-added hydrocarbons.
1 Introduction
Benzene, toluene and xylenes (BTXs), ethylbenzene, indenes and naphthalenes are generally extracted from petroleum, and widely utilized in the petrochemical industry as feedstock for plastics, pesticides, transportation fuels, synthetic fibers and chemicals.1,2 With the decrease in fossil fuels and the increase in energy consumption, lignocellulosic biomass will be used as an alternative to replace the fossil feedstock in the future. Nowadays, the use of biomass derived from agricultural residues has been widely accepted for the production of biofuel and biogas worldwide due to its low cost and eco-friendliness. Sunflower stalks are one abundant agricultural biomass residue which can be considered a huge energy source. However, to date, this kind of biomass residue still lacks an alternative use. In particular, by pyrolysing this biomass, a large amount of bio-oil containing various oxygenated chemicals such as ketones, aldehydes, furans, acids, and phenolic compounds will be generated. It is possible to selectively convert these oxygenated compounds to high value-added hydrocarbons such as BTXs and others via catalytic upgrading processes.3–5 To date, many researchers have used zeolites to upgrade bio-oil via cracking, cyclization, oligomerization and aromatization due to their excellent shape selectivity and controllable acidity.6–8 However, the amount of hydrocarbon in the upgraded bio-oil obtained using traditional zeolites is not that high due to the limitation of the open pore size. Kelkar et al.9 reported that those oxygenated compounds with a large molecular size derived from lignin have difficulty diffusing into the small pores of zeolites and other microporous solid catalysts, and cracked on the surface of the catalysts rapidly, leading to rapid deactivation of the catalyst and a low hydrocarbon yield.To solve the reactant diffusion problem in the pores, mesoporous catalysts such as Al-MCM-41 and Al-MSU-S have been developed.9–11 Yu et al.12 found that the use of La-Al-MCM-41 can reduce the amount of oxygenated compounds in bio-oil by up to 43.1%. Park et al.13 upgraded bio-oil over mesoporous MFI zeolite with a pore size of about 5 nm, and found that this kind of catalyst had a high selectivity for BTXs production. Mesoporous alumina should be another suitable catalyst for deoxygenation reactions due to its high acidity and large surface area.14 However, only a few studies have investigated the effect of pore size of Al2O3 or metal modified Al2O3 for the selective catalytic upgrading of bio-oil to aromatic hydrocarbons. On the other hand, Al2O3 prepared using a conventional hydrothermal synthesis without template assistance always has a low surface area and broad pore size distribution, which will affect the selectivity and activity of the obtained Al2O3. In contrast, by using a surfactant-templating method, it is possible to achieve a high surface area and uniform pores.15
Pluronic P123 (PEO)20(PPO)70(PEO)20, a commercially available triblock copolymer is an interesting surfactant template for mesoporous alumina synthesis as it is inexpensive and biodegradable.16 The textural properties of alumina such as its surface area and morphology can be controlled by using it in a synthesis process. For aromatic hydrocarbon production from the upgrading of bio-oil, a pore size range of about 3–9 nm is generally required, which could be realized by adjusting the P123/Al molar ratio. On the other hand, it should be noted that severe coking usually occurs on pure Al2O3 due to excessive dehydrogenation reactions at the Lewis acid sites.17 Modification by loading a transition metal on Al2O3 or other porous materials can improve the acidity and generate new acid sites which are beneficial for the selective production of hydrocarbons from the oxygenated compounds in bio-oil and the resistance to coke deposition of the catalyst.18–21
In this study, mesoporous alumina with a controllable pore size distribution and high surface area was synthesized firstly using a P123-assisted hydrothermal method and then, copper (Cu) and iron (Fe) were separately doped on it with different loading amounts. The prepared catalysts were characterized using BET surface area measurements, X-ray diffraction (XRD), scanning electron microscopy coupled with an energy dispersive X-ray detector (SEM-EDX), H2-temperature programmed reduction (H2-TPR) and NH3-temperature programmed desorption (NH3-TPD) analysis. The Cu or Fe loaded mesoporous alumina with optimum loading amount was determined by using the prepared catalysts for the in situ deoxygenation of bio-oil derived from the fast pyrolysis of sunflower stalks. The reusability of the catalyst with the best performance was also investigated both with and without regeneration. A catalytic system with high effectiveness and long-term stability was expected to be obtained to convert the oxygenated compounds in bio-oil to high value-added hydrocarbons.
2 Experimental
2.1 Biomass feedstock
Sunflower stalks (Aomori, Japan) were crushed and sieved with a size range of 1–2.8 mm and dried at 105 °C in an oven. Their proximate, ultimate and ash compositions were reported in our previous work.222.2 Synthesis of mesoporous alumina
Various mesoporous Al2O3 powders were prepared using a Pluronic P123 surfactant-assisted hydrothermal synthesis method with different P123/Al molar ratios from 0.005 to 0.05. In brief, 10.72 g of Al(NO3)3·9H2O (Wako, Japan) was dissolved in 36 mL of deionized water with vigorous stirring and then 9.06 g of urea (Wako, Japan) was added to it with continuous stirring at 35 °C for 30 min. After complete dissolution, a certain amount of P123 (Aldrich, Germany) was introduced into the solution with continuous stirring until a homogeneous solution was achieved. Thereafter, the mixture was transferred to a Teflon-lined stainless steel autoclave with a volume of 50 mL and heated at 120 °C for 24 h. After the reaction, the white precipitate was separated, washed with deionized water, dried at 105 °C in oven and then calcined at 750 °C in air for 2 h. Here, the resultant product is denoted as Al2O3-X, where X represents the molar ratio of P123/Al.2.3 Preparation of transition metal-modified Al2O3 catalyst
1, 2.5, 5 and 10 wt% of Cu were separately loaded on the prepared mesoporous Al2O3 using an impregnation method. Firstly, 1 g of Al2O3 powder was added into 20 mL of Cu(NO3)2·3H2O (Wako, Japan) aqueous solution and stirred at room temperature for 2 h. Then, the obtained slurry was dried at 80 °C and subsequently calcined at 650 °C in air for 2 h. Meanwhile, Fe (1, 2.5, 5 and 10 wt%) loaded on Al2O3 was also prepared using the same method. Here, the prepared catalyst is denoted as Me/Al2O3, where Me is Cu or Fe.2.4 Catalyst characterization
N2 adsorption–desorption conducted at −196 °C using a Quantachrome instrument (NOVA 4200e, USA) was performed to investigate the specific surface area, pore volume and pore size distribution (BJH method) of the catalysts. Prior to N2 sorption measurement, the catalyst sample was degassed under vacuum at 200 °C for 2 h. The XRD patterns were determined using an X-ray diffractometer (XRD, Rigaku Smartlab, Japan) in the 2θ range of 30–90° with a scanning step of 0.02° using Cu Kα radiation (λ = 0.1542 nm). The morphology of the catalyst and the existence of alkali and alkaline earth metal (AAEM) species on the spent catalyst were investigated using scanning electron microscopy (SEM, SU8010, Hitachi, Japan) coupled with an energy dispersive X-ray detector (EDX). The reduction behavior of the metal loaded Al2O3 was carried out using H2 temperature-programmed reduction (H2-TPR) with a BET-CAT catalyst analyzer (BEL, Japan). The H2 consumption of each sample was monitored with a TCD detector from room temperature to 750 °C with a heating rate of 5 °C min−1 under a 5% H2/Ar steam with a total flow rate of 50 cm3 min−1. The acidity and acid site distribution of the catalyst were investigated using NH3 temperature-programmed desorption (NH3-TPD) with the same instrument as the H2-TPR analysis. The catalyst was saturated with NH3 steam at ambient temperature with a total flow rate of 50 cm3 min−1 for 1 h and then purged with He steam at 120 °C for 2 h to remove physisorbed NH3 in the catalyst structure. After stabilization, NH3 desorption was carried out from room temperature to 800 °C with a heating rate of 10 °C min−1 under a 50 cm3 min−1 flow of He. Here, the NH3 desorption peak was also detected using a thermal conductivity detector (TCD) and the adsorbed NH3 concentration was quantified from the peak area by calibration using the standard gas. Before H2-TPR and NH3-TPD measurements, all samples were preheated at 750 °C for 1 h under He flow to remove moisture and some impurities within the catalyst structure. Temperature-programmed oxidation (TPO) of the coke deposited on the spent catalyst was conducted using a thermogravimetric analyzer (TGA, DTG-60H, Shimadzu, Japan). The sample was pretreated at 120 °C for 1 h prior to measurement and then heated at a heating rate of 10 °C min−1 until a temperature of 700 °C was achieved under a flow of air.2.5 Catalytic deoxygenation of bio-oil
A schematic diagram of the experimental setup for the in situ catalytic deoxygenation of bio-oil derived from the fast pyrolysis of biomass has been reported elsewhere.23 In a typical run, 0.1 g of biomass particles and 0.8 g of catalyst were separated with quartz wool and packed in a fixed bed reactor. During the reaction, N2 gas flow (100 cm3 min−1) was used as the carrier gas. Prior to the experiment, the reactor was purged with a N2 gas flow for about 10 min to remove the air inside. The fast pyrolysis reaction temperature, reaction time and heating rate were fixed at 565 °C, 4 min and 1000 °C min−1, respectively, which are the optimum conditions to obtain the highest bio-oil yield from our preliminary experiments. The liquid product was trapped using two acetone ice-cooling bottles and the non-condensed gas was passed over a CaCl2 filter and collected in a gas bag for further analysis.2.6 Analysis of bio-oil and gas products
The obtained bio-oil was analyzed using gas chromatography (GC-2010 Plus, Shimadzu, Japan)/mass spectrometry (GCMS-QP2010 Ultra, Shimadzu, Japan) with an Ultra ALLOY+ 5 capillary column. The bio-oil samples were automatically injected into the column whose temperature was increased from 50 to 300 °C with a ramp rate 10 °C min−1 and maintained at 300 °C for 10 min. The ionization chamber of the MS setup was set at 200 °C. Various peaks in the chromatogram corresponding to various compounds such as aromatics, aliphatics, ketones, phenols, sugars, acids, aldehydes, furans and others were identified by comparing with the built-in NIST spectral library. Here, the products with boiling points lower than 300 °C could be detected. In addition, the peak area percentages obtained from the GC-MS chromatogram were considered to be a good estimation because they relate to the concentration of the main products in bio-oil. The water content in the bio-oil was measured using a Karl-Fisher titration method (MKS-500, KEM, Japan). The collected non-condensed gas was analyzed off-line using gas chromatography (Agilent 7890A GC system, USA) equipped with a thermal conductivity detector (TCD) and 3 packed columns (1 molecular sieve 5A column + 1 HayeSep Q column + 1 molecular sieve 5A column) to separate CO, CH4 and CO2 using He as the carrier gas, while a molecular sieve 5A for H2 detection was used with Ar as a carrier gas.3 Results and discussion
3.1 Physicochemical properties of the prepared catalysts
Fig. 1A shows the N2 adsorption–desorption isotherms of mesoporous alumina prepared with different molar ratios of P123/Al. Here, all the mesoporous alumina have IUPAC type VI isotherms, which correspond to mesoporous materials.24 The steep capillary condensation step at 0.4–0.8 P/P0 indicates a high degree of mesoporous uniformity. It should be noted that the broader capillary condensation step of Al2O3-0.05 indicates a decrease in uniformity of the sample.16 For Al2O3 without P123 assistance, its isotherm exhibits a hysteresis loop shape, which is classified as type H3 with slit-shaped pores. Fig. 1B shows the pore-size distributions of the prepared mesoporous Al2O3. Here, Al2O3-0.005, Al2O3-0.01 and Al2O3-0.025 exhibit narrow pore size distributions with a pore size range of 4–10 nm. In contrast, Al2O3 and Al2O3-0.05 have broader pore size distributions with a larger pore size range of 3–20 nm. The specific surface area, pore volume and pore size of these mesoporous Al2O3 are summarized in Table 1. It can be seen that the pore volumes and pore sizes of Al2O3 increase with increasing P123/Al molar ratio from 0.005 to 0.05 while the surface area continuously increases until 0.01, indicating that the concentration of P123 has a great effect on the textural properties of the obtained Al2O3. Such a large pore size can allow the compounds with larger molecules in bio-oil to diffuse within the pores. However, it should be noted that a high P123 concentration in the initial solution could affect the self-assembly of P123 molecules to form mesoporous Al2O3 as expected. As shown in Table 1, for the transition metal loaded Al2O3-0.01, the surface area, pore volume and pore size are significantly reduced since the metal species could deposit in the internal and external mesopores of Al2O3 and change the surface properties. When the metal loading amount is increased to 10 wt%, the pore size is greatly decreased. This should be due to pore blockage and metal sintering in the catalyst structure.25 | ||
| Fig. 1 (A) N2 adsorption–desorption isotherms and (B) pore-size distributions of Al2O3, Al2O3-0.005, Al2O3-0.01, Al2O3-0.025 and Al2O3-0.05. | ||
| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| Al2O3 | 195 | 0.22 | 2.29 |
| Al2O3-0.005 | 285 | 0.51 | 4.19 |
| Al2O3-0.01 | 345 | 0.79 | 5.81 |
| Al2O3-0.025 | 327 | 0.94 | 6.86 |
| Al2O3-0.05 | 311 | 0.96 | 8.42 |
| 1 wt% Cu/Al2O3-0.01 | 332 | 0.78 | 5.80 |
| 2.5 wt% Cu/Al2O3-0.01 | 315 | 0.75 | 5.77 |
| 5 wt% Cu/Al2O3-0.01 | 302 | 0.71 | 5.72 |
| 10 wt% Cu/Al2O3-0.01 | 309 | 0.69 | 5.70 |
| 1 wt% Fe/Al2O3-0.01 | 339 | 0.78 | 5.80 |
| 2.5 wt% Fe/Al2O3-0.01 | 333 | 0.76 | 5.79 |
| 5 wt% Fe/Al2O3-0.01 | 326 | 0.75 | 5.76 |
| 10 wt% Fe/Al2O3-0.01 | 317 | 0.70 | 5.71 |
Fig. 2 shows the XRD patterns of pure Al2O3-0.01 without metal loading and of various metal-loaded Al2O3-0.01 catalysts. One can see that in the cases of low metal loading (1–5 wt%), no diffraction peaks corresponding to metal loaded Al2O3 can be observed compared with pure Al2O3-0.01 without metal loading, indicating that the metal particles with a small size have been well dispersed on the mesoporous Al2O3 without accumulation during the preparation process.26 In contrast, at high metal loading (e.g., 10 wt% of Cu or Fe), peaks corresponding to copper oxide or iron oxide phases can be clearly observed, indicating the accumulation and/or bad dispersion of large metal particles. These XRD analysis results are also in good agreement with the BET analysis.
 | ||
| Fig. 2 XRD patterns of (A) 1–10 wt% Cu/Al2O3-0.01 and (B) 1–10 wt% Fe/Al2O3-0.01. | ||
Fig. 3 shows the SEM images of the various catalysts. As shown in Fig. 3A and B, the mesoporous Al2O3 prepared with P123 assistance has a rod-like morphology with a uniform larger particle size compared to Al2O3 without P123 assistance, which shows a broken-sheep-wool-like morphology.
 | ||
| Fig. 3 SEM images of (A) Al2O3, (B) Al2O3-0.01, (C) 2.5 wt% Cu/Al2O3-0.01, (D) 10 wt% Cu/Al2O3-0.01, (E) 2.5 wt% Fe/Al2O3-0.01 and (F) 10 wt% Fe/Al2O3-0.01. | ||
During the mesoporous Al2O3 synthesis, numerous ammonium aluminum carbonate hydroxide (AACH) crystals can be formed gradually with the increase in pH via urea decomposition. As such, –OH groups of AACH crystals should be further adsorbed on the oxide groups of P123 surfactant micelles via hydrogen bonding, leading to the systematic formation of mesoporous rod-like particles. As shown in Fig. 3C–F, when 2.5 wt% Cu or Fe metal is loaded on Al2O3-0.01, no metal bulk can be observed and the surface of the rods remains smooth. However, in the case of high Cu or Fe loading (10 wt%), the surface becomes hairy, especially in the case of Cu loading.
Fig. 4 shows the H2-TPR profiles of the various catalysts. For 1–10 wt% Cu/Al2O3-0.01, shown in Fig. 4A, at the low temperature range, the main reduction peak of the CuO species corresponding to the conjunct reductions from Cu2+ to Cu+ and from Cu+ to Cu0 can be clearly observed. Here, the small reduction peak appearing at high temperature is ascribed to the reduction of the CuAl2O4 phase.27 Furthermore, it should be noted that the reduction peaks shift towards the lower temperature range when the Cu loading amount is increased. This could be a result of the interaction between the Al2O3 support and Cu species with different particle sizes. In general, at higher Cu loadings (e.g. 10 wt% Cu loading), larger particles would be formed, resulting in a weaker interaction due to the poorer dispersion of Cu species. As a result, a lower temperature is needed for the reduction by hydrogen.28 On the other hand, for 1–10 wt% Fe/Al2O3-0.01, as shown in Fig. 4B, the reduction peaks are present, especially for 5 and 10 wt% Fe loading, which reveals the isolated reduction step of Fe2O3 at low temperature and of FeAl2O4 at high temperatures as follows:27
| Fe2O3 + H2 → Fe3O4 + H2O | (1) |
| Fe3O4 + H2 → Fe0 + H2O | (2) |
| FeAl2O4 + H2 → Al2O3 + Fex2+O | (3) |
| Fex2+O + H2 → Fe0 + H2O | (4) |
 | ||
| Fig. 4 H2-TPR profiles of (A) 1–10 wt% Cu/Al2O3-0.01 and (B) 1–10 wt% Fe/Al2O3-0.01. | ||
Here, Fe/Al2O3 has the opposite trend in the H2-TPR profiles compared to Cu/Al2O3 with increasing metal loading. It is possible that they have different ionic radii, different interactions with Al2O3 and different microstructure on mesoporous Al2O3.
Fig. 5 shows the NH3-TPD profiles of various catalysts. Here, a single NH3-desorption peak in a wide temperature range of 150–500 °C is observed in the pure Al2O3 without metal loading. After Cu or Fe metal is loaded on Al2O3, the desorption peak area increases to a wider temperature range, indicating that the substitution of proton sites with the doping metal species occurs, resulting in the generation of new proton sites on the catalyst.29 Moreover, two NH3 desorption peaks corresponding to the weak and strong acid sites appear for the 2.5 to 10 wt% Cu or Fe loaded Al2O3, indicating the formation of new strong acid sites due to the metal loading. It should be noted that the amount of weak acid sites also decreases with the increase in strong acid sites when the metal loading amount is increased. Table 2 shows the acidities quantified from the peak area for these catalysts. One can see that the acidity is increased after metal loading. However, the acidity gradually decreases with increasing metal loading, which could result from the covering of some proton sites with larger metal particles.30 Here, the high acidity should be beneficial for deoxygenation and aromatization, leading to the reduction of oxygenated compounds in bio-oil with the increase in aromatic hydrocarbons; on the other hand, if the acidity is too high, coke is more easily formed due to the cracking of hydrocarbons on the catalyst, resulting in rapid deactivation during the deoxygenation process.
 | ||
| Fig. 5 NH3-TPD profiles of (A) 1–10 wt% Cu/Al2O3-0.01 and (B) 1–10 wt% Fe/Al2O3-0.01. | ||
| Catalyst | Acidity (mmol g−1, low temp.) | Acidity (mmol g−1, high temp.) | Total acidity (mmol g−1) |
|---|---|---|---|
| Al2O3-0.01 | 0.271 | — | 0.271 |
| 1 wt% Cu/Al2O3-0.01 | 0.602 | — | 0.602 |
| 2.5 wt% Cu/Al2O3-0.01 | 0.530 | 0.054 | 0.584 |
| 5 wt% Cu/Al2O3-0.01 | 0.357 | 0.074 | 0.431 |
| 10 wt% Cu/Al2O3-0.01 | 0.295 | 0.102 | 0.397 |
| 1 wt% Fe/Al2O3-0.01 | 0.630 | — | 0.630 |
| 2.5 wt% Fe/Al2O3-0.01 | 0.500 | 0.007 | 0.507 |
| 5 wt% Fe/Al2O3-0.01 | 0.415 | 0.010 | 0.425 |
| 10 wt% Fe/Al2O3-0.01 | 0.438 | 0.009 | 0.447 |
3.2 Deoxygenation of bio-oil over the prepared catalysts
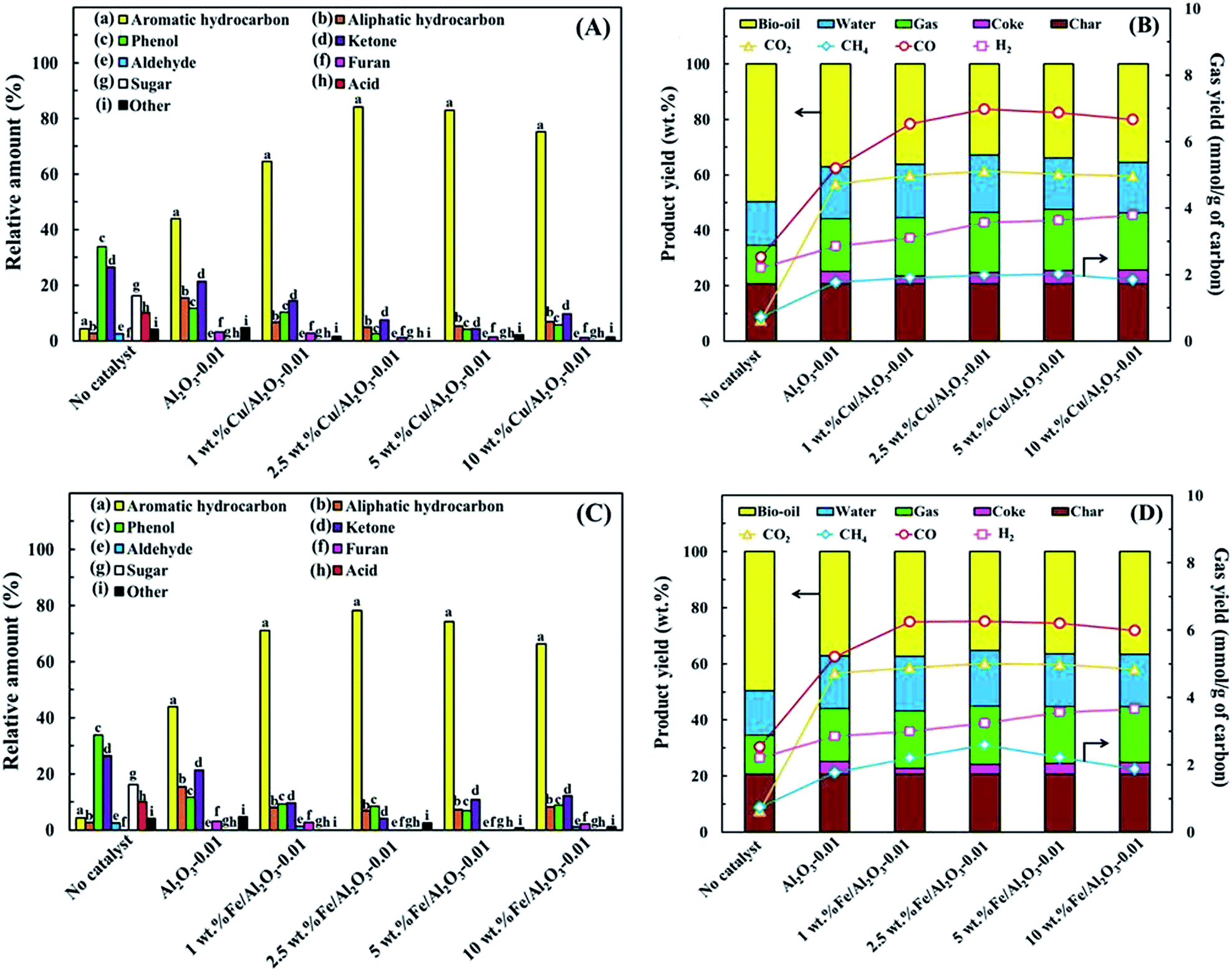
In this study, the chemicals in the bio-oil products detected using GC-MS analysis are classified into nine groups, i.e., aromatic hydrocarbons, aliphatic hydrocarbons, phenols, ketones, aldehydes, furans, sugars, acids and others. Among these, the relative total amounts of aromatic and aliphatic hydrocarbons in the upgraded bio-oil are used as indicators to evaluate the deoxygenation efficiency of each catalyst. Fig. 6A shows the relative amount of the chemical composition of the bio-oils in the absence and presence of various Al2O3-X catalysts. One can see that the amount of oxygenated compounds such as acids, anhydrosugars and phenolic compounds are significantly reduced in the presence of catalysts, resulting in an obvious increase in the relative total hydrocarbon amount. The reduction of phenolic compounds such as phenols and cresols can be attributed to the occurrence of dehydration and dihydroxylation; the decrease in acidic components results from decarboxylation and decarboxylation over the catalyst; the sugars are converted to furans at first via a dehydration reaction, and further converted to allene via decarbonylation.20,31 Here, allene is one of the main precursors for aromatic hydrocarbon production by oligomerization. From Fig. 6A, one can see that even when pure Al2O3 prepared without P123 assistance is used, the total relative hydrocarbon amount in the bio-oil also increases by up to 46% compared to the case without catalyst. Furthermore, the relative total hydrocarbon amount is also found to be more increased with increasing surface area and pore size in mesoporous Al2O3, indicating that the large molecules in bio-oil can easily diffuse into the catalyst pores and contact with more active sites so that they can be easily converted to hydrocarbons. A maximum relative total hydrocarbon amount of 59% is obtained using Al2O3-0.01. In contrast, for Al2O3-0.025 and Al2O3-0.05, the relative total hydrocarbon amount is lower than that using Al2O3-0.005 and Al2O3-0.01. This demonstrates that mesoporous Al2O3-0.01 with a pore size of about 5.8 nm is more suitable for deoxygenation, especially for aromatic hydrocarbon production. | ||
| Fig. 6 (A) Chemical composition and (B) aromatic selectivity of the upgraded bio-oils obtained from the in situ catalytic deoxygenation of bio-oil derived from fast pyrolysis of sunflower stalks using various Al2O3-X with different initial P123/Al molar ratios. | ||
Fig. 6B shows the distribution of aromatic hydrocarbons in the upgraded bio-oils using various Al2O3-X. Here, the aromatic hydrocarbons are categorized as MAHs, including benzene, toluene, xylenes and ethylbenzene, and PAHs including indenes and naphthalenes. It is found that different pore sizes of the Al2O3 catalysts lead to different selectivity for aromatic hydrocarbon production. As shown in Fig. 6B, the highest BTXs selectivity of 59% is obtained using Al2O3 prepared without P123 assistance. With increasing pore size of the catalyst, the MAHs amount in the upgraded bio-oil decreases while the PAHs amount clearly increases. This indicates that the pore size of Al2O3 plays a significant role in the aromatic selectivity of the upgraded bio-oil. However, even though the Al2O3-0.01 catalyst has the highest catalytic activity compared with the others, PAHs as undesirable products are still high in the case when pure mesoporous Al2O3 is used. It has been reported that although mesoporous catalysts with large pores are beneficial for the diffusion of compounds with large molecular sizes, it also promotes PAH formation, more easily leading to coking on the catalyst via condensation and polymerization reactions.9
To minimize the amount of PAHs and promote MAH formation, Cu or Fe loaded Al2O3-0.01 with various loadings were tested for the deoxygenation of bio-oil. As shown in Fig. 7A, the relative total hydrocarbon amount increased from 59 to 71.3% using 1 wt% Cu/Al2O3-0.01 instead of Al2O3-0.01, and further increased to 89% using 2.5 wt% Cu/Al2O3-0.01. Here, oxygenated compounds such as phenols and ketones in the upgraded bio-oil are significantly reduced. This confirms that the loading of Cu on Al2O3 can greatly promote dehydroxylation, dehydration and decarbonylation reactions. Interestingly, the main hydrocarbons in the upgraded bio-oil using Cu/Al2O3-0.01 are aromatic hydrocarbons, indicating that this catalyst favors promoting the conversion of aliphatic to aromatic. It is possible that the existence of Cu on Al2O3 can increase the Lewis acid sites or electron pair acceptors and promote hydride ion release, which is beneficial for the transformation of olefins to carbenium ions through intermediate dienes, and substantially enhances aromatic formation.32 In addition, a possible route for aromatic hydrocarbon formation in the in situ catalytic upgrading of bio-oil derived from biomass pyrolysis is shown in Fig. 8 and can be described as follows: firstly, cellulose and hemicellulose are decomposed to anhydrosugars such as levoglucosan (LGA), 1,6-anhydro-β-D-glucofuranose (AGF) and levoglucosenone (LGO) via a pyrolysis process. These sugars then undergo dehydration and re-arrangement reactions to form furans. The furans are carried by the carrier gas to pass through the catalyst layer, where they undergo decarbonylation to form C2–C4 alkenes or alkynes in the mesopores. Finally, the formed olefins are converted to cyclic aliphatic hydrocarbons via combination and further transform to aromatic hydrocarbons via Diels–Alder reactions and cyclization aromatization.2
 | ||
| Fig. 7 (A, C) Chemical compositions and (B, D) mass balance of the products obtained from the in situ catalytic upgrading of bio-oil derived from the fast pyrolysis of sunflower stalks using the various catalysts. | ||
 | ||
| Fig. 8 The possible reaction mechanism for aromatic hydrocarbon formation obtained from the in situ catalytic deoxygenation of bio-oil derived from the pyrolysis of biomass. | ||
The lignin in biomass is decomposed to phenols and phenol alkoxy species via depolymerization at first and then the aromatics can be formed by decarbonylation, dehydration and dehydroxylation. Meanwhile, char is generally formed via re-polymerization and secondary pyrolysis of biomass. However, when the loading amount of Cu is further increased from 5 to 10 wt%, the hydrocarbon amount decreases while oxygenated compounds increase to some extent. It is possible that the occurrence of metal sintering results in the reduction of acidity. Moreover, the decrease in surface area and pore size also influences the mass transfer. For Fe loaded Al2O3-0.01, as shown in Fig. 7C, the same trend as Cu/Al2O3-0.01 for deoxygenation is found. However, Fe loaded catalysts show a lower catalytic activity. As indicated by the NH3-TPR results, more proton sites and new strong acid sites are available in the Cu/Al2O3-0.01 catalysts than Fe/Al2O3-0.01 for aromatization, resulting in more aromatic hydrocarbon formation.
Fig. 7B and D show the mass balance of the products and gas yields derived from the in situ catalytic upgrading of bio-oil using 1–10 wt% Cu/Al2O3-0.01 and 1–10 wt% Fe/Al2O3-0.01. One can see that the yield of bio-oil clearly decreases while the gas yield increases compared to the case without catalyst. Here, deoxygenation reactions such as dehydration, decarboxylation, decarbonylation, oligomerization, cracking, aromatization and dehydrogenation during the catalytic upgrading process result in an increase of the yields of CO, CO2, H2O and coke. Considering the coke formation, when 1 wt% Cu or Fe loaded Al2O3-0.01 is used to replace Al2O3-0.01 to upgrade bio-oil, the yield of coke clearly decreases. This should be attributed to the promotion of hydrogen atom migration through C–H activation on the catalytically active sites.33,34 However, higher metal loading leads to an increase in coke deposition due to the catalytic decomposition of the gas phase and polycondensation of furans, phenols and aromatics on the metal species.
Fig. 9 shows the distribution of aromatic hydrocarbons in the upgraded bio-oil using 1–10 wt% Cu/Al2O3-0.01 and 1–10 wt% Fe/Al2O3-0.01. It can be seen that a high selectivity towards MAHs over 80% can be obtained in the presence of 1–2.5 wt% Cu or Fe/Al2O3-0.01. In contrast, when the loading amount is over 2.5 wt%, more PAHs are generated. This indicates that overloading of Cu or Fe on Al2O3 promotes secondary reactions, favoring the formation of aromatics with larger molecular sizes such as naphthalenes via further aromatization and polymerization. Moreover, overloading of Cu or Fe on Al2O3 may also favor alkylation with MAHs to form PAHs.
 | ||
| Fig. 9 Aromatic selectivity in the upgraded bio-oils obtained from the in situ catalytic deoxygenation of bio-oil derived from fast pyrolysis of sunflower stalks using various 1–10 wt% metal/Al2O3-0.01 catalysts. | ||
Fig. 10 shows the effect of the catalyst amount on the deoxygenation efficiency based on the relative total hydrocarbon amount in the upgraded bio-oil using 2.5 wt% Cu/Al2O3-0.01 and 2.5 wt% Fe/Al2O3-0.01. Here, the biomass amount in the reactor is fixed but the catalyst amount is adjusted to change the residence time of the reactants in the catalyst layer. One can see that the yield of bio-oil decreases when the catalyst/biomass weight ratio increases since the deoxygenation reactions could occur on more active sites, which results in the generation of more gas, water, coke and others. In this study, an optimum catalyst/biomass weight ratio of 8 is obtained using 2.5 wt% Cu/Al2O3-0.01 as well as 2.5 wt% Fe/Al2O3-0.01. However, when the weight ratio is increased over 8, a little reduction in the relative total hydrocarbon amount is observed. This is probably due to the promotion of secondary reactions, where hydrocarbon molecules are further cracked into gases, coke and others during the long residence time.
 | ||
| Fig. 10 Effect of catalyst amount on the relative total hydrocarbon amount in the upgraded bio-oil obtained using (A) 2.5 wt% Cu/Al2O3-0.01 and (B) 2.5 wt% Fe/Al2O3-0.01. | ||
3.3 Catalyst reusability
Fig. 11 shows the reusability of 2.5 wt% Cu/Al2O3-0.01 and 2.5 wt% Fe/Al2O3-0.01 for the in situ deoxygenation of bio-oil. In this study, both catalysts were tested for 5 cycles in the same operation conditions, in which from the first to the fourth cycle, the spent catalyst in each cycle was reused without regeneration, and in the fifth cycle, it was regenerated by their calcination at 650 °C for 30 min in air before testing. One can see that both catalysts show an increase in the relative total hydrocarbon amount at the first reuse cycle compared with the fresh ones. This indicates that the reused catalysts have better catalytic activity to promote deoxygenation.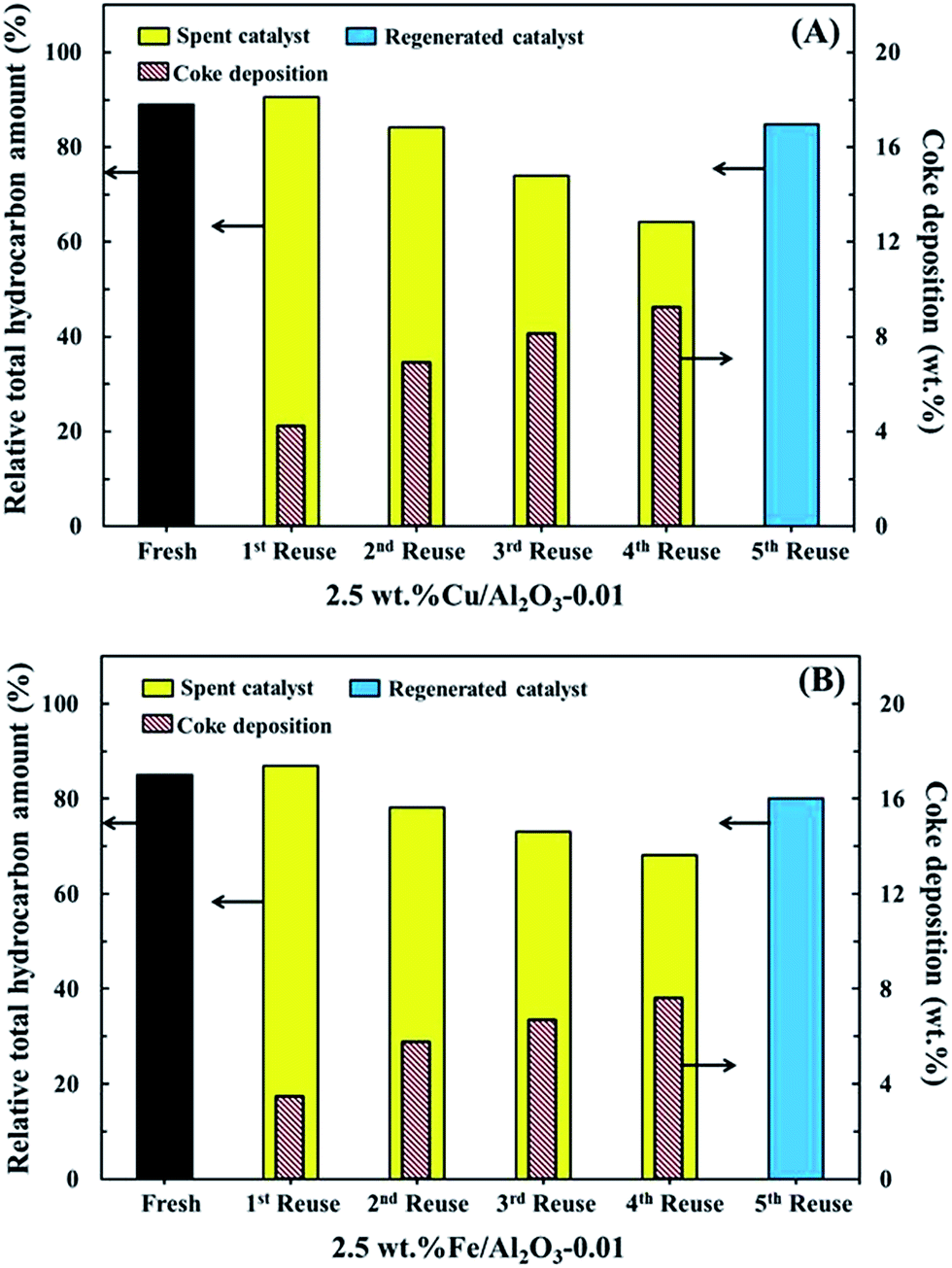 | ||
| Fig. 11 Reusability and regeneration performance of (A) 2.5 wt% Cu/Al2O3-0.01 and (B) 2.5 wt% Fe/Al2O3-0.01 for the in situ catalytic deoxygenation of bio-oil derived from the fast pyrolysis of sunflower stalks. | ||
The effect of alkali metal or alkaline earth metal (AAEM) species such as potassium from the biomass deposited on the spent catalyst should also have a contribution.35 In the presence of the in situ produced H2O and AAEM, more H2 can be generated during biomass pyrolysis and the deoxygenation process via a redox cycle reaction (Fig. 12). Here, the in situ produced H2 may enhance the dehydrogenation pathway on the catalyst, resulting in the increase of alkanes as a source of MAHs. Also, when additional H2 is present during the deoxygenation process, reactive H˙ radicals can be generated, which can simultaneously promote oxygen removal via capping free radicals.18 In order to confirm the presence of AAEM species, element mapping of the spent catalysts was performed and the results are shown in Fig. 13. It can be clearly seen that K element exists on the surface of the catalyst. Here, K should be the main AAEM species because its content in the ash of sunflower stalks is found to be up to 72%. However, the relative total hydrocarbon amount gradually reduces after the first reuse cycle. This should be a result of the increased coke deposition on the catalyst with increasing test cycle (Fig. 10). Interestingly, one can see that 2.5 wt% Fe/Al2O3-0.01 without any regeneration has better long-term stability with a reduction in the relative total hydrocarbon amount of 19% after the fourth reuse cycle compared to 2.5 wt% Cu/Al2O3-0.01 (26%). This difference can be attributed to the coke amount formed on the catalyst.
 | ||
| Fig. 12 The overall reaction of AAEM-catalyzed pyrolysis of biomass. M is the AAEM species. | ||
 | ||
| Fig. 13 SEM images, EDX mapping images and EDX spectra of (A–D) 2.5 wt% Cu/Al2O3-0.01 and (E–H) 2.5 wt% Fe/Al2O3-0.01 after reaction (4th reuse). | ||
Fig. 14 shows the TPO signals of the spent catalysts (after 4th reuse), which represent the thermal decomposition of deposited coke on the catalyst. One can see that Cu/Al2O3 and Fe/Al2O3 have different thermal decomposition ranges. In the case of Cu/Al2O3, two thermal decomposition ranges can be observed for coke burning.
 | ||
| Fig. 14 TPO profiles of the spent catalysts after reaction (4th reuse). | ||
In contrast, for Fe/Al2O3, only one low thermal decomposition range is observed. This indicates that the formed coke has different types and/or particle sizes. It has been reported that coke with a lower removal temperature range belongs to oxygenated coke while that with a higher removal temperature range is graphite-like coke.34 Furthermore, the coke amount on 2.5 wt% Cu/Al2O3-0.01 after the 4th reuse is 9.2%, which is higher than that on 2.5 wt% Fe/Al2O3-0.01 (7.6%). It is also possible that 2.5 wt% Cu/Al2O3-0.01 has a higher acidity than 2.5 wt% Fe/Al2O3-0.01. In addition, the new strong acid sites of 2.5 wt% Cu/Al2O3-0.01 might promote the generation of stable graphite-like coke on the catalyst. On the other hand, after both catalysts are regenerated, i.e., the fifth reuse cycle, no serious reduction in the relative total hydrocarbon amount was found when compared with the fresh catalysts, indicating that the performance of the spent catalyst can be perfectly recovered using this regeneration method.
4 Conclusions
Cu and Fe loaded uniform rod-like mesoporous alumina synthesized using a P123-assisted hydrothermal method were usied for the in situ deoxygenation of bio-oil derived from fast biomass pyrolysis in a fixed bed reactor. It is found that Cu or Fe loaded mesoporous Al2O3 with a loading amount in the range of 1–2.5 wt% with a good metal species dispersion showed a high catalytic activity for the selective catalytic deoxygenation of bio-oil to MAHs. At higher metal loading conditions, the metal species was found to be accumulated in the mesoporous Al2O3 structure, resulting in a lower deoxygenation ability. Mesoporous Al2O3 with a large pore size resulted in an increased PAHs content in the upgraded bio-oil. 2.5 wt% Cu/Al2O3-0.01 gave the highest relative total hydrocarbon amount of 89%. AAEM species in the biomass were found to be carried to the surface of the catalysts during the pyrolysis process and promote the deoxygenation reactions in the deoxygenation process, which led to an increased relative total hydrocarbon amount in the upgraded bio-oil. Also, the deactivated spent catalysts were easily regenerated by calcination in air. It is expected that such catalysts can be widely used for the deoxygenation of bio-oil in practical processes.Acknowledgements
This study was supported by Aomori City Government, Japan, and the International Joint Research Project of Shanxi Province (No. 2015081051 and 2015081052), China. S. Karnjanakom, and A. Bayu gratefully acknowledges the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan for the scholarship.References
- A. Zheng, Z. Zhao, S. Chang, Z. Huang, H. Wu and X. Wang, J. Mol. Catal. A: Chem., 2014, 383–384, 23–30 CrossRef CAS.
- G. Li, L. Yan, R. Zhao and F. Li, Fuel, 2014, 130, 154–159 CrossRef CAS.
- W. Liu, K. Tian, H. Jiang, X. Zhang, H. Ding and H. Yu, Environ. Sci. Technol., 2012, 46, 7849–7856 CrossRef CAS PubMed.
- Z. Ma, W. Zhou, L. Wei, L. Jia, B. Hou, D. Li and Y. Zhao, RSC Adv., 2015, 5, 88287–88297 RSC.
- M. Asadieraghi, W. M. A. W. Daud and H. F. Abbas, RSC Adv., 2015, 5, 22234–22255 RSC.
- C. A. Mullen, A. A. Boateng, R. B. Dadson and F. M. Hashem, Energy Fuels, 2014, 28, 7014–7024 CrossRef CAS.
- B. Zhang, Z. P. Zhong, X. B. Wang, K. Ding and Z. W. Song, Fuel Process. Technol., 2015, 138, 430–434 CrossRef CAS.
- T. Mochizuki, S. Y. Chen, M. Toba and Y. Yoshimura, Appl. Catal., A, 2013, 456, 174–181 CrossRef CAS.
- S. Kelkar, C. M. Saffron, K. Andreassi, Z. Li, A. Murkute, D. J. Miller, T. J. Pinnavaia and R. M. Kriegel, Appl. Catal., B, 2015, 174–175, 85–95 CrossRef CAS.
- M. H. Nilsen, E. Antonakou, A. Bouzga, A. Lappas, K. Mathisen and M. Stöcker, Microporous Mesoporous Mater., 2007, 105, 189–203 CrossRef CAS.
- G. T. Neumann and J. C. Hicks, ACS Catal., 2012, 2, 642–646 CrossRef CAS.
- F. Yu, L. Gao, W. Wang, G. Zhang and J. Ji, J. Anal. Appl. Pyrolysis, 2013, 104, 325–329 CrossRef CAS.
- H. J. Park, H. S. Heo, J. K. Jeon, J. Kim, R. Ryoo, K. E. Jeong and Y. K. Park, Appl. Catal., B, 2010, 95, 365–373 CrossRef CAS.
- T. S. Nguyen, S. He, L. Lefferts, G. Brem and K. Seshan, Catal. Today, 2016, 259, 381–387 CrossRef CAS.
- P. Bai, F. Su, P. Wu, L. Wang, F. Y. Lee, L. Lv, Z. Yan and X. S. Zhao, Langmuir, 2007, 23, 4599–4605 CrossRef CAS PubMed.
- K. L. Materna, S. M. Grant and M. Jaroniec, ACS Appl. Mater. Interfaces, 2012, 4, 3738–3744 CAS.
- R. Feng, S. Liu, P. Bai, K. Qiao and Y. Wang, J. Phys. Chem. C, 2014, 118, 6226–6234 CAS.
- F. Melligan, M. H. B. Hayes, W. Kwapinski and J. J. Leahy, Energy Fuels, 2012, 26, 6080–6090 CrossRef CAS.
- A. Veses, B. Puértolas, J. M. López, M. S. Callén, B. Solsona and T. García, ACS Sustainable Chem. Eng., 2016, 4, 1653–1660 CrossRef CAS.
- Y. T. Cheng, J. Jae, J. Shi, W. Fan and G. W. Huber, Angew. Chem., Int. Ed., 2012, 51, 1387–1390 CrossRef CAS PubMed.
- A. Veses, B. Puértolas, M. S. Callén and T. García, Microporous Mesoporous Mater., 2015, 209, 189–196 CrossRef CAS.
- S. Karnjanakom, G. Guan, B. Asep, X. Du, X. Hao, C. Samart and A. Abudula, Energy Convers. Manage., 2015, 98, 359–368 CrossRef CAS.
- S. Karnjanakom, G. Guan, B. Asep, X. Hao, S. Kongparakul, C. Samart and A. Abudula, J. Phys. Chem. C, 2016, 120, 3396–3407 CAS.
- B. Huang, C. H. Bartholomew, S. J. Smith and B. F. Woodfield, Microporous Mesoporous Mater., 2013, 165, 70–78 CrossRef CAS.
- C. Wu, L. Dong, J. Onwudili, P. T. Williams and J. Huang, ACS Sustainable Chem. Eng., 2013, 1, 1083–1091 CrossRef CAS.
- A. Alihosseinzadeh, B. Nematollahi, M. Rezaei and E. N. Lay, Int. J. Hydrogen Energy, 2015, 40, 1809–1819 CrossRef CAS.
- A. H. M. Batista, F. S. O. Ramos, T. P. Braga, C. L. Lima, F. F. Sousa, E. B. D. Barros, J. M. Filho, A. S. Oliveira, J. R. Sousa, A. Valentini and A. C. Oliveira, Appl. Catal., A, 2010, 382, 148–157 CrossRef CAS.
- Y. Zhu, X. Kong, X. Li, G. Ding, Y. Zhu and Y. Li, ACS Catal., 2014, 4, 3612–3620 CrossRef CAS.
- W. Liu, H. Wang, A. M. Karim, J. Sun and Y. Wang, Chem. Soc. Rev., 2014, 43, 7594–7623 RSC.
- D. Gao, H. Yin, A. Wang, L. Shen and S. Liu, J. Ind. Eng. Chem., 2015, 26, 322–332 CrossRef CAS.
- E. F. Iliopoulou, S. D. Stefanidis, K. G. Kalogiannis, A. Delimitis, A. A. Lappas and K. S. Triantafyllidis, Appl. Catal., B, 2012, 127, 281–290 CrossRef CAS.
- S. Kelkar, C. M. Saffron, Z. Li, S. S. Kim, T. J. Pinnavaia, D. J. Miller and R. Kriegel, Green Chem., 2014, 16, 803–812 RSC.
- L. Wang, H. Lei, Q. Bu, S. Ren, Y. Wei, L. Zhu, X. Zhang, Y. Liu, G. Yadavalli, J. Lee, S. Chen and J. Tang, Fuel, 2014, 129, 78–85 CrossRef CAS.
- W. L. Fanchiang and Y. C. Lin, Appl. Catal., A, 2012, 419–420, 102–110 CrossRef CAS.
- J. Rizkiana, G. Guan, W. B Widayatno, X. Hao, X. Li, W. Huang and A. Abudula, Appl. Energy, 2014, 133, 282–288 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |