
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhodium-doped titania photocatalysts with two-step bandgap excitation by visible light—influence of the dopant concentration on photosensitization efficiency†
J.
Kuncewicz
*a and
B.
Ohtani
b
aFaculty of Chemistry, Jagiellonian University, ul. R. Ingardena 3, 30-011 Kraków, Poland. E-mail: kuncewic@chemia.uj.edu.pl; Fax: +48 126340515; Tel: +48 126632005
bInstitute for Catalysis, Hokkaido University, Sapporo 001-0021, Japan. E-mail: ohtani@cat.hokudai.ac.jp; Fax: +81 11 706 9133; Tel: +81 11 706 9132
First published on 15th August 2016
Abstract
Development of photocatalysts active under visible light has been achieved through the modification of titania with rhodium (Rh) ions. Depending on the concentration of the precursor of Rh species, various crystalline phases and modification modes have been obtained. Lower Rh concentrations (below 0.5%) resulted in doped rutile particles, while higher (above 1%) gave mainly surface modified anatase particles. High photocatalytic activities were obtained for surface enriched doped rutile-titania with an extremely low amount of trivalent rhodium (Rh3+) ions (about 0.0005 mol%). Almost four orders of magnitude higher Rh concentration (2%) also provided highly active materials, however, they were composed of mainly anatase particles with photoactive surface RhOx species. Extremely low Rh concentration allowed to limit the amount of intrinsically present tetravalent rhodium (Rh4+) species, which may decrease charge mobility in the titania lattice and promote the recombination route in Rh-doped materials. Photoactive Rh species created in low concentrated samples were able to drive reversible redox processes controlled by the range of light energies. Photocatalytic activity tests, action spectra and photoelectrochemical measurements allowed to propose a unique photosensitization mechanism, involving an Rh3+/Rh4+ redox couple acting as built-in redox mediator, in which both electrons in the conduction band (CB) and positive holes in the valence band (VB) might be generated upon irradiation with visible light. Such a mechanism was not valid for materials containing higher amounts of Rh composed of mainly anatase. Detailed studies on the properties of the materials containing various amounts of Rh species (0.0001–3 mol%) showed the dependency of efficiency of photoinduced processes on Rh concentration.
Introduction
Development of visible-light active inorganic materials able to drive simultaneously redox processes that require high oxidation and reduction potentials, such as oxidation of water and reduction of oxygen, is still facing problems concerning charge-carrier generation efficiency and stability. There are only a few bare semiconductors that possess high oxidation as well as reduction power in the excited state, such as titanium(IV) oxide (titania), strontium titanate, cadmium sulfide, zinc sulfide or zirconium oxide, moreover, most of them are unstable or photocatalytically inactive under visible light.1 Photosensitization of suitable wide band-gap semiconductors usually does not solve the problem. Commonly used methods for the activation of semiconductors toward visible range of light, such as dye sensitization or doping with metal or non-metal ions,2–6 seem to provide materials maintaining only one of the intrinsic oxidative or reductive properties. In such materials it is impossible to induce one-step band-gap excitation only with visible-light irradiation. To overcome this problem and provide simultaneous generation of highly active holes and electrons, a complex z-scheme-resembling systems, in which reduction and oxidation processes have been targeted to two different semiconductors, have been proposed.7 Activity of such systems is based on the electron-transfer reaction between particles of two materials with anodic and cathodic characters, mediated with an external redox pair or carried out without any redox mediators.8 Nevertheless, z-scheme photocatalysis has many drawbacks arisen from complexity of the systems and limited efficiency of interfacial electron transfer between particles of different origins. Thus, another concept for developing efficient visible-light active systems might evolve from the introduction of the idea of z-scheme mechanism into a single photocatalyst system. It should be possible if a suitable inner redox mediator would be introduced to (or combined with) semiconductor particles of proper redox potentials of conduction and valence-band edges.Such idea should be feasible in doped materials, where new electronic states are introduced into the band gap of base material. Titania, due to its redox abilities, seems to be a good candidate for development of such single photocatalyst system. There are many transition metal ions, e.g. Mo, Co, Cr, Fe 6,9,10 that may efficiently reduce the bandgap of titania leading to the red shift of the optical absorption edge, however, only a few are able to induce appearance of intermediate bands which divide the energy gap into sub-gaps.9 According to computational calculations from group of 4d transition metals such behavior should be shown by Tc, Ru, Rh and Pd ions.11–13
Rh-doped titania has been already suggested as an efficient photocatalyst for O2 evolution under visible light.14 It is evidenced that introduction of Rh ions to the rutile lattice, similarly as in Rh-doped SrTiO3-based materials, induces substitution of Ti(IV) ions with guest species and creation of new electronic states located within the band gap of titania.14–17 The electronic structure of doped materials depends strongly on the valency of introduced ions. Due to the charge compensation insertion of Rh3+ induces Rh4+ species and oxygen vacancies generation. Theoretical studies, focused on the materials containing about 1.4% of Rh, show that in such conditions the donor states just at the top of VB associated with Rh3+ ions and interband-states arising from the presence of Rh4+, which may act as acceptors of electrons from VB, should be created.13 However, in Rh-doped titania, as in the case of titania modified with Rh3+ species located only at the surface,18–20 visible-light-induced activity is assigned mainly to the excitation of electron from Rh3+ to the CB of semiconductor, which is followed by the chemical reduction of generated Rh4+. Same mechanism was presented by Niishiro et al. for 1% Rh-doped SrTiO3.15,16 Although the authors shown that part of intrinsically present Rh4+ species may be photoreduced with electrons from the VB in the initial step of photocatalytic process, it is believed that consequent irradiation of the photocatalytic system induces mainly excitation of Rh3+. Photo-regenerated in the initial step Rh3+ ions are regarded as “unstable” compared with intrinsically present Rh3+ species stabilized with oxygen vacancies.15
In the case of both, titania and SrTiO3 based Rh-doped materials, the possibility of photogeneration of Rh4+ from intrinsically present Rh3+ and the role of photoreduction of Rh4+ in over whole photocatalytic process have not been investigated. Moreover, Rh4+ ions formed in the doping process are believed to play a role of recombination centers, what was a main reason of photocatalytic inactivity of as prepared Rh-doped SrTiO3,15,21 and Rh-doped titania containing 1 mol% of Rh.14 Thus, up to now, to restore photoactive Rh3+ species in Rh4+ reach-materials or to prevent the creation of Rh4+ species two strategies have been used: reduction with hydrogen flow, and, more successful, codoping with ions of different valency (e.g. Sb5+) allowing charge compensation within the host lattice.14,15,21 However, total reduction of Rh4+ ions within the lattice and stabilization of Rh3+ species provided by second strategy results in the disappearance of available interband states,13,14 what was calculated and indirectly observed as bleaching of absorption band associated with Rh4+ in codoped samples, and put the possibility of visible-light-induced excitation of valence band electrons into question.
Our approach is to decrease the number of recombination centres while maintaining the pot of “unstable” Rh3+ species by decreasing the concentration of the dopant to the extremely low value. As it was studied by Bloh et al. effective concentration of dopant is strongly dependent on particles size, and may reach very small values.22,23 If the amount of Rh3+ ions is low enough and they are enriching the surface of the semiconductor particles the charge compensation by Rh4+ generation should be partly suppressed. Such conditions should allowed creation of Rh3+ ions which are able to drive reversible redox processes. In these processes photogeneration of stable Rh4+ ions as well as their photoreduction should be possible. Very low amounts of introduced impurities should have also less impact on the decrease of charges mobility which was suggested for the codoped materials.21–24 Recently our studies on such “depleted” Rh-doped titania have shown that photogenerated Rh4+ species play important role in overall photocatalytic activity.25 We developed materials presenting unique photosensitisation mechanism assuring two-step bandgap excitation involving Rh3+/Rh4+ coupled redox pair. However, still the influence of the intrinsic Rh3+ and Rh4+ concentration on the efficiency of photoinduced processes was not well understood.
In the present work we investigated the influence of Rh concentration on properties of the materials and photosensitization mechanism by detailed studies on the morphology, crystalline structure, photoabsorption, photocatalytic and photoelectrochemical properties of materials containing broad range of the Rh concentration: from 0.00001 to 3 mol%. We propose the best conditions for the highest efficiency of two-step bandgap excitation, and discuss future perspectives of improvement of the performance of visible-light-active materials acting by such mechanism.
Results and discussion
The Rh–TiO2 materials have been obtained by impregnation of titania (anatase) with RhCl3 and calcination at 700 °C for 3 h. The calcination conditions were sufficient for RhCl3 decomposition. The concentration of Rh significantly influenced the crystalline structure of host material. XRD analysis revealed that materials containing up to 0.1% of Rh had a single phase of rutile. Additionally, no changes in the lattice constants could be observed for less concentrated samples. It may indicate not efficient incorporation of Rh ions into the host lattice or, more likely, too small changes in the lattice parameters to be detected due to low Rh concentration. Further increase of Rh content caused enlargement of rutile lattice constants, what suggests that Rh ions are introduced to rutile crystalline lattice in more concentrated samples (Fig. 1S in ESI†). Though, it is hard to conclude if there are any changes in anatase phase parameters. Together with an increasing Rh concentration retardation of anatase–rutile transformation could be also observed (Fig. 1), and the samples containing 2% were composed mainly of anatase particles. Moreover, for all samples there is no evidence of any other crystalline phases (e.g. Rh2O3) occurring in studied materials.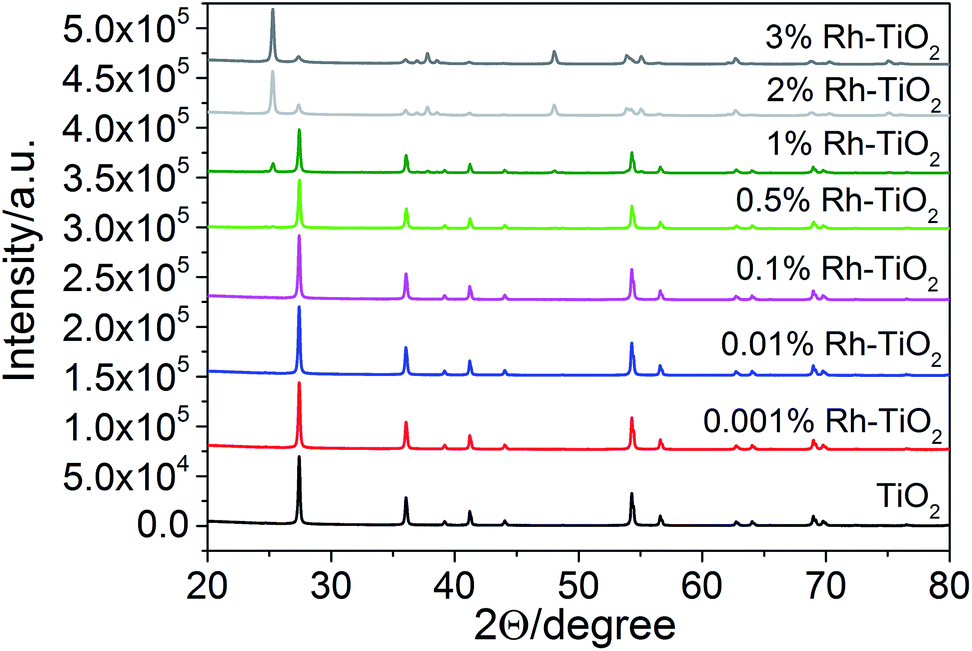 | ||
| Fig. 1 XRD patterns of Rh–TiO2 and a reference sample without rhodium. | ||
Particle size of obtained materials depended on their crystalline structure. Samples containing mainly rutile phase were characterized of particles with an average diameter included within the range 70–120 nm. With an increase of anatase-phase content a slight decrease in particles size has been observed. The changes in particle size estimated on the basis of SEM and TEM images (Fig. 2 and 3) were in agreement with the changes in surface area which was estimated to 11.8 m2 g−1 for bare titania and 10.2, 10.0, 17.3, 24.2 for 0.001%, 0.05%, 1%, and 3% Rh–TiO2, respectively. TEM images (Fig. 3) and STEM/EDX analysis (Fig. 4) revelled also that in the case of low concentrated photocatalysts (up to 0.5% of Rh) composed of mainly rutile phase the materials are homogenous and Rh ions are uniformly located within all of the particles. No surface significant accumulation or localized aggregation of Rh species could be observed. The materials containing higher Rh concentration and anatase content are characterized of inhomogeneous Rh distribution. On the basis of the size and resistance of the observed particles to laser ablation during the measurement, it is suggested that Rh species accumulated at the anatase particles.
 | ||
| Fig. 2 SEM images of Rh–TiO2 materials containing 0% (a), 0.001% (b), 0.01% (c), 0.1% (d), 0.5% (e), 1% (f), 2% (g) and 3% (h) Rh. | ||
 | ||
| Fig. 3 TEM images of Rh–TiO2 materials containing 0% (a), 0.001% (b), 0.5% (c), 1% (d), 2% (e), 3% (f) of Rh. | ||
 | ||
| Fig. 4 STEM images (left) and Rh signal from EDX analysis (right) of 0.5% (a) and 2% (b) Rh–TiO materials. | ||
The valency of Ti and Rh species located at the surface and sub-surface area of studied materials has been investigated by XPS measurements. Oxidation states were determined by the binding energies of Rh 3d5/2 and Ti 2p3/2.
Samples with concentration of Rh up to 0.5% contained overlapped signals about 309.0 eV and 309.8 eV, assigned to Rh3+ and Rh4+, respectively (Fig. 5 left).26,27 The Rh4+/Rh3+ ratio was increasing with Rh concentration reaching the highest value for 1% Rh–TiO2. Together with that change a slight shift of the binding energy-value of Rh 3d5/2 signal, especially Rh4+ component (from 309.87 eV to 309.68 eV), toward lower energies has been observed. It may indicate enrichment of the surface with doped Rh4+ ions that should change their surroundings. Decomposition of RhCl3 in air results in rhodium oxide generation and Rh2O3 is more in the creation of oxides agglomerates characterized mainly by Rh3+ signal. Thus, the presence of Rh4+ species may indicate introduction of Rh ions into the titania lattice, at least close to the surface. Above 1% of Rh the ratio Rh4+/Rh3+ decreased and signal of Rh3+ became predominant, what indicates competition between incorporation and agglomeration processes at the titania surface in such a conditions. What is more, the broadening of the signal recorded for highly concentrated samples could be observed. Deconvolution of the spectra revealed that new component appeared at ar. 308.2 eV (Fig. 6) what may be associated with Rh3+ species with different surroundings, such as in Rh2O3 agglomerates.27,28 The concentration of such species located at the surface of titania increased gradually with amount of Rh. Insignificant change in the ratio of Rh4+ to Rh3+ primary signals in samples containing more than 1% of Rh clearly shows that mainly new species were generated above that concentration. The observed change correlated with an increase of anatase structure contribution with Rh content. The analysis of Rh valency in materials containing less than 0.5% of Rh was insufficiently reliable due to the detection limits of used technique, which was around 0.1%.
 | ||
| Fig. 5 XPS spectra of a Rh–TiO2 materials with various Rh-concentration: Rh(3d5/2) (left), and Ti 2p3/2 (normalized) (right) peaks. | ||
 | ||
| Fig. 6 Deconvolution of the Rh(3d5/2) signals recorded for 0.5% (a), 1% (b), 2% (c) 3% (d). | ||
Titanium present at or near to the surface of bare titania gave peak of the energy around 458.45 eV assigned to 2p3/2 of Ti4+ (Fig. 5 right).17 Increase of Rh concentration in material caused progressive shift of the binding energy of Ti 2p3/2 signal up to 457.96 ± 0.03 eV, which was observed for 1% and higher concentrations of Rh. It may indicate changes in Ti ions surrounding and Fermi level shifts caused by doping process.17 For materials containing more than 2% of Rh new component of Ti 2p3/2 signal at binding energies around 456.9 eV was observed, which might suggest possible contribution of Ti3+ ions.
Ratio of Rh/Ti calculated from XPS data is higher than the nominal one, what indicates that the surface is enriched with Rh ions.
Obtained samples kept in the dark were characterized of pale yellow to orange colour which was reversibly turning to sage tinge upon day light irradiation. Samples containing more than 1% of Rh became more brownish and did not change the colour under the light. Intensity of the colours was gradually increasing with Rh concentration.
DRS absorption spectra of the samples containing 0.0001–0.001% of Rh comprise characteristic for titania band-gap-absorption accompanied with a weak absorption with an offset at 550–590 nm assigned to Rh3+ strongly interacting with TiO2 (Fig. 7 inside).14,17 For samples with more than 0.005% of Rh two bands within visible range of light can be distinguished. More intensive one at 445 nm and broad weak band (resembling tailoring for less concentrated samples) at ar. 620 nm associated with Rh3+ and Rh4+ species strongly interacting with titania lattice, respectively. The former one results from the electron transition from Rh3+ ion to the CB of titania, and the latter one originates from the electron transition from VB of titania to Rh4+ ion.14,17 The observed absorption does not resemble the absorption spectra of bare Rh2O3 and a 0.01% mixture of Rh2O3 with titania (Fig. 2S in ESI†), what indicates, that does not originate from the rhodium oxide microcrystalline species. The changes observed for the latter material after calcination at 700 °C may suggest partial introduction of Rh ions into the crystalline lattice of titania in a such prepared sample.
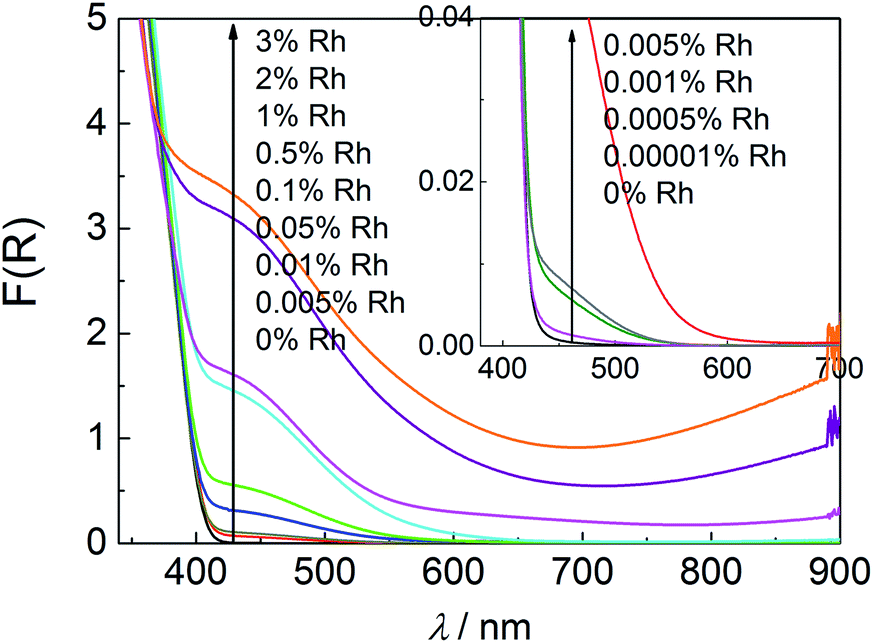 | ||
| Fig. 7 Diffuse reflectance spectra of Rh–TiO2 with various Rh-concentration and a reference sample without Rh species. | ||
Samples containing more than 0.5% of Rh are characterised of strong absorption within whole range of visible light increasing with the Rh content and, in result, covering characteristic band at 620 nm in the case of 2% and 3% Rh–TiO2. It suggests creation of new types of Rh species at the surface of doped titania particles. Observed increased absorption above 750 nm may be assigned to d–d charge transfer transition between Rh3+ and Rh4+ species, as it was suggested for Rh-doped SrTiO3.29,30
Light induced photoabsorption changes
Materials with lower Rh concentration show unusual spectroscopic behavior under exposure to light. Similarly to the changes observed for the previously studied 0.01% Rh–TiO2 material (calcined at 650 °C)25 irradiation with light from the range overlapping the band assigned to Rh3+ (440–550 nm) caused decrease of the intensity of that band and increase of the absorption intensity of band at. 620 nm assigned to Rh4+ (Fig. 8 left). The process was reversed through the irradiation with light of the range 590–730 nm, covering the range of the Rh4+ band. It is clear that former irradiation causes electron transition from Rh3+ to the CB of titania. The latter one should induce photo reduction of generated Rh4+ by the electron from the VB.14,17 | ||
| Fig. 8 Diffuse reflectance spectra and differential spectra (inside) in the representation of Kubelka Munk function (F(R)) of 0.01% Rh–TiO2 material kept in the dark and irradiated with the light in the ranges: 440–730 nm, and 590–730 nm (left). Changes in the value of F(R) observed at 620 nm for Rh–TiO2 materials with various Rh-concentration induced by the irradiation of the samples with light in the range 440–730 nm (right). The changes were calculated in respect to the F(R) values recorded for samples irradiated with low-energy light (590–730 nm). | ||
The photoreduction of Rh4+ has been observed previously in Rh-doped SrTiO3, however only in the induction period of photocatalytic reaction in which just intrinsically present Rh4+ ions participated.15,16 Due to that in such materials two kinds of Rh3+ ions have been distinguished: Rh3+ stabilized by oxygen vacancies created during the doping process, and Rh3+ “unstable”, which may be created from intrinsically present Rh4+ by the photoreduction process at the beginning of photocatalytic reaction. However, in published papers there was little discussion on fate of Rh3+ ions participating in photoexcitation process, and no information about photoreduction process of Rh4+ ions created through the photooxidation of Rh3+ species have been provided. Results of our studies indicate that concentration of “unstable” Rh3+ ions capable of reversible photoredox processes in the studied materials is much higher than concentration of Rh3+ ions just created upon reduction of intrinsically present Rh4+ (included into calculations). As it was shown before,25 only a very little fraction of Rh3+ species participating in reversible redox processes, is oxidized to Rh4+ in air in the dark. Most of such Rh3+ ions are stable in the presence of O2 and the only oxidizing factor is light (Fig. 8 left).
Such a light induced photoabsorption changes were observed for the samples containing from 0.001% up to 1% of Rh. The analysis of the absorption changes recorded for the samples containing less than 0.001% of Rh was difficult due to the very small ratio of the observed differences in the KM function values to the noise.
The concentration of reversibly oxidized Rh3+ ions increased with Rh concentration reaching the maximum for samples with 0.5% of Rh species (Fig. 8 right).25 For samples containing 1% of Rh the amount of photogenerated Rh4+ slightly decreased. Increase of Rh concentration caused also extension of the time necessary for total Rh3+ recovery in air, which became significant for samples containing more than 0.01%. Moreover, the range of the red light efficiently inducing recovery of Rh3+ was slightly shifted toward lower energies for more concentrated samples. The materials prepared by calcination of the mixture of Rh2O3 and TiO2 did not show any significant light-induced absorption changes (Fig. 3S in ESI†), as well as visible light-photocatalytic activity, what indicates importance of the strong interaction of Rh ions with titania lattice in these processes.
Materials with higher Rh concentration (2 and 3%) did not present such spectroscopic behaviour. In those samples UV-vis light as well as red light caused only very slight decrease in the absorption intensity within whole range of visible light. The reason of observed changes is still under considerations.
Photocatalytic and photoelectrochemical activity
 | ||
| Fig. 9 Rates of CO2 liberation in first two hours of the process of acetaldehyde oxidative decomposition in the presence of Rh–TiO2 with various Rh concentrations irradiated with visible light (λ > 440 nm). A horizontal line shows CO2-generation rate in the presence of 0% Rh–TiO2. | ||
Since in the samples with lower Rh concentration two types of dependent on each other photoinduced redox processes can be distinguished, the photocatalytic activity should result from the efficiency of both processes.
As it was shown in our previous paper, transient Rh3+/Rh4+ ratio in irradiated samples in air depends strongly on the wavelength range of incident light.25 Red shift of the used range of visible light causes increase of the ratio (Fig. 4Sa in ESI†). Similar dependency was observed in a course of photocatalytic reaction, however in the presence of organic electron donor (AcH) in the system photoreduction of Rh4+ was enhanced which was explained by improved consumption of holes generated within VB of titania (Fig. 4Sb in ESI†).25 Nevertheless, if the range of light was limited to 440–550 nm significant decrease of Rh3+/Rh4+ was observed. Such changes indicated that photoreduction of Rh4+ is one of the main photoinduced processes occurring not only in the initial step but also in a coarse of the photocatalytic process conducted under whole range of visible light. It was shown also, that decrease of Rh4+ concentration during the photocatalytic reaction may be achieved by utilization of light characterized of proper wavelength ranges. Such procedure should improve the efficiency of over whole photocatalytic process through the elimination of recombination centers together with the creation of VB holes.
The evidence of the importance of Rh4+ photoreduction process has been also shown in double beam photocatalytic activity measurements.25 Simultaneous irradiation of the 0.01% Rh–TiO2 (650 °C) material with two light ranges: 440–550 nm – inducing the electron transfer from Rh3+ to titania CB, and 590–730 nm – initiating the excitation of the electrons from the VB to Rh4+, gave synergetic effect which was not observed for bare titania (Fig. 5S in ESI†). This experiment indicated also that induction of the electron transfer from Rh3+ to CB of titania is essential for the occurrence of the process of Rh4+ photoreduction. Such coupling of these two processes is confirmed by the results of action spectra and photoelectrochemical measurements performed for the applied potentials from the range 0.5–1.0 V (vs. Ag/AgCl). The experiments showed that monochromatic light induces only the photoexcitation of Rh3+ (Fig. 10, Fig. 11 left). Similar results of action spectra measurements were obtained for 1% Rh-doped SrTiO3.16 It was suggested that obtained dependency of the apparent quantum yield of photocatalytic reaction on the energy of monochromatic light proved the photosensitization mechanism based mainly of the Rh3+-CB transition. Considering occurrence of both dependent on each other photoinduced processes in a coarse of photocatalytic reaction the analysis of results of the experiments performed with monochromatic light should be careful.
 | ||
| Fig. 10 Photocurrent as a function of wavelength of incident light recorded in aerated 0.1 M KNO3 for Rh–TiO2 materials with various Rh-concentration (left and right). Applied potential: 0.95 V vs. Ag/AgCl. | ||
 | ||
| Fig. 11 Action spectra together with absorption spectra recorded for bare TiO2, and 0.001% Rh–TiO2 (left), and 2% Rh–TiO2 (right) in the reaction of oxidative decomposition of AcH in air. | ||
According to obtained results it might be expected that materials with higher content of Rh containing higher concentration of Rh3+ able to drive reversible redox processes will show higher photocatalytic activity under visible light. However, increase of Rh concentration caused progressive deactivation of the materials. It might be explained by two phenomena: hindrance of the absorption of titania within its band edge caused by overlaid Rh species located at the surface or negative impact of doped ions to charges mobility in titania CB and enhanced recombination.22,23 The results of photoelectrochemical measurements clearly showed that the titania-originated photoactivity induced by light from the range of bandgap absorption decreased with Rh concentration, even in the case of extremely small amount of Rh (Fig. 10 left), what indicates validity of the latter assumption. What is more, the photoelectrochemical activity within the range of visible light resulting only from the efficiency of Rh3+ excitation increased with Rh content. Similarly, the photocatalytic activity tests performed under the light from the range limited to 550–730 nm, under which bare titania shows negligible activity, showed improvement of photocatalytic activity of materials with an increase of Rh-concentration (Fig. 12). Both improvements were observed up to 0.1% and 0.01% of Rh, respectively. For higher concentrations, up to 1%, both titania- and Rh-originated activity decreased, as it might be seen in the Fig. 10 (left).
 | ||
| Fig. 12 Amounts of CO2 and AcH monitored during process of AcH oxidative decomposition in the presence of Rh–TiO2 with various Rh-concentration under irradiation with light of the range 550–730 nm. | ||
Materials containing more than 1% of Rh revealed different features. The action spectra measurements performed for 2% Rh–TiO2 have shown that maximal efficiency of the photocatalytic process was obtained upon irradiation with 515 nm light (Fig. 11, right). Taking into account also a lack of the noticeable amount of Rh3+ ions able to carry on reversible redox reactions and the results of XPS measurements, improvement of the visible light-induced photocatalytic activity of more concentrated materials should result from the activity of different type of surface Rh species compared with low concentrated samples. The significance of surface character of the modification is proved by the high value of photocurrents in the spectral range of titania absorption resulting from low efficiency of recombination processes (Fig. 10 right), what suggests negligible concentration of Rh ions within the lattice of anatase. Thus, the formation of surface species seems to be favorable for particles with anatase crystalline structure, which is dominant in more concentrated samples. It is in agreement with previous studies showing that Rh-doping process is more preferential in the case of rutile than anatase.12 What is more, rutile phase seems to be essential for the creation of Rh3+ ions able for reversible redox reactions. Low visible light-induced electrochemical activity observed for 2% Rh–TiO2 compared with strongly improved photocatalytic activity in that light range suggests that created surface species may also partially cause an improvement of separation of photogenerated charges. However, the complicity of the composition of highly concentrated samples significantly impedes mechanistic studies of observed processes and the exact mechanism of visible light-induced activity of highly concentrated materials was not fully examined.
Location of new electronic states on the scale of potentials – spectroelectrochemical measurements
Spectroelectrochemical measurements performed in a broad range of the applied potentials allowed to study the redox potential of Rh3+ species in less concentrated samples. During the experiments samples were electrochemically oxidized and changes in the reflectance at 625 nm, reflecting the presence of Rh4+ species, have been recorded. The potential of CB edge of studied materials have been studied by following the changes in the reflectance at 780 nm during negative potential application.The consecutive oxidation of the samples containing from 0.01 to 0.5% of Rh resulted in the increase of the absorption at 625 nm (Fig. 13, left). In the case of all samples potential ar. 0.9–1.1 V vs. Ag/AgNO3 electrode (depending on the amount of Rh), at which the beginning of the linear increase in KM function was observed, could be distinguished. It may indicate the position of electronic states created upon modification on the scale of potential. The magnitudes of changes in KM function value corresponded to concentration of generated Rh4+ ions. The reference experiments in which spectra of 0.5% Rh–TiO2 and bare titania have been recorded at 0.5, 1.0, and 1.5 V of applied potential clearly showed that appearing absorption originated from Rh4+ species (Fig. 14 left). It resembles absorption which appeared during the irradiation of the Rh-modified samples with white light (Fig. 8 left).
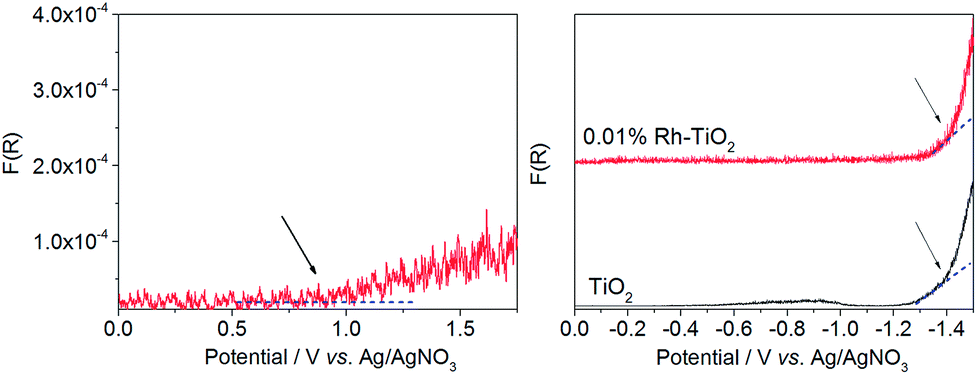 | ||
| Fig. 13 Changes of KM function at 625 nm (left) and 780 nm (right) with the applied potential recorded for 0.5% Rh–TiO2 (left) and 0.01% Rh–TiO2 and bare TiO2 (right) deposited at the surface of platinum plate. The measurements were carried out in deoxygenated 0.1 mol dm−3 LiClO4 solution in acetonitrile. | ||
 | ||
| Fig. 14 Reflectance spectra in the representation of KM function recorded for 0.5% Rh–TiO2 (left) and bare titania (right) deposited at the surface of platinum plate at the various applied potentials (E vs. Ag/AgNO3). The measurements were carried out in deoxygenated 0.1 mol dm−3 LiClO4 solution in acetonitrile. | ||
The results of studies on the redox properties of titania (both modified and bare) indicate that the potential of conduction band edge is around −1.4 V vs. Ag/AgNO3 electrode regardless of the type of the sample (Fig. 13, right). Comparison of the potentials of the conduction band edge, additional states created upon modification with Rh and optically determined band gap energies (ca. 3.0 eV for all samples) draw to conclusion that acceptor electronic states are located within the band gap of titania (Fig. 15).
 | ||
| Fig. 15 Scheme of the bands structure on the scale of potentials and possible light-induced electron transfer processes presented for Rh–TiO2. | ||
Location of photoactive species in low-concentrated samples
The location of Rh ions strongly depends on the Rh-concentration. The characterisation of materials containing extremely low amount of Rh was very hard due to limitations of used methods, thus the nature of the modification was deduced from the tendency observed in the case of more concentrated samples.Used preparation method should have assured uniform distribution of the Rh species, even in the case of very low Rh contents. The lack of aggregation of Rh species during the preparation was indicated by the linear increase of the absorption band within visible range of light with Rh concentration observed for impregnated but not calcined samples with extremely low Rh concentration (Fig. 6S in ESI†). The STEM/EDX analysis (Fig. 4) also proved homogenous distribution of Rh ions within the particles for more concentrated materials. What is more, the results of XRD measurements performed for materials with higher Rh concentrations indicated that the used method allowed to introduce Rh ions into the rutile lattice. Moreover, XPS analysis indicated enrichment of the surface area with Rh ions. Thus, it is concluded that Rh-species in low-concentrated samples should strongly interact with titania lattice, however doping process may occur mainly in the surface area of the titania particles. It is expected that the depth of penetration of Rh-ions increases with Rh-content, thus for extremely low concentrated samples the ratio of Rh doped species located at the surface to the species located in the bulk should be the highest. Doping process was also indicated by the light induced absorption changes observed for studied materials. Such reversible redox processes controlled by light occurred only in the samples in which photogenerated Rh4+ ions were stable, and were not observed in the case of anatase-based materials, where doping process is less favorable, nor in the case of materials prepared by mixing titania with rhodium(III) oxide. What is more, such changes were also not reported for titania grafted with Rh3+ ions, in which rhodium(III) oxide species are located at the surface of semiconductor.20
In the case of materials with extremely low Rh-concentration such surface located Rh-species appeared to be the main photoactive species responsible for the improvement of the photocatalytic activity under visible light irradiation. Photoabsorption and photocatalytic activity measurements performed for these materials kept in water at 90 °C, rinsed four times with water and dried at room temperature in air showed that such intensive rinsing caused slight depletion of the absorption bands in the visible range of light accompanied with large decrease of photocatalytic activity within same range of radiation (Fig. 7S in ESI†). The moderate changes in the intensity of the Rh-originated absorption band upon rinsing may indicate that other, less active, Rh species are doped more deeply in the bulk of titania. The influence of the type of Rh species on titania properties is still under investigations.
Conclusions
The obtained Rh-modified titania showed visible-light activity towards oxidative decomposition of AcH. The most active materials appeared at 0.0005% and 2% of Rh concentrations. The obtained results indicate that lower Rh concentrations (below 0.5%) resulted in doped rutile particles, while higher (above 0.5%) gave mainly surface modified anatase particles. Due to the complicity of the composition of more concentrated samples and great advantage from economical point of view of extremely low doping ratio, the mechanistic studies were focused mainly on the low concentrated materials.Low Rh concentration allowed creation of Rh species able to drive reversible photo-induced redox processes controlled by the range of light energies. Such species play crucial role in the unique photosensitization mechanism revealed for studied materials. It is shown that photosensitization mechanism suggested for materials with extremely low Rh content is based on two-step bandgap excitation, in which excitation of the electrons from Rh3+ to the CB is followed by the light induced-electron transfer from the VB to photogenerated Rh4+. What is more, the mechanism assures not only generation of both electrons and holes within titania bands but also recovery of photoactive Rh3+ ions during the photocatalytic process.
It was found that essential condition for the formation of such a Rh species that can induce photoinduced reversible redox reactions is rutile structure of host material. In the case of more concentrated doped materials (2%, 3% Rh–TiO2) anatase–rutile transformation was retarded, which caused the formation of materials with different Rh species and photosensitization mechanism.
In the case of rutile-based materials increase of the amount of photoactive Rh species capable of reversible redox reactions could be achieved by an increase of nominal Rh content, which was confirmed by the photoabsorption, photocatalytic activity and photoelectrochemical measurements. The efficiency of the visible-light-induced Rh-originated activity increased with Rh content reaching the maximum at about 0.01–0.1% of Rh. However, increase of Rh concentration simultaneously caused enhancement of recombination and/or deterioration of charges mobility within titania bands. Introduction of extremely low amount of Rh allowed limit the amount of intrinsically present tetravalent rhodium (Rh4+) species, which is believed to decrease charge mobility in titania lattice and promote recombination route in Rh-doped materials. It probably contributed also to the limitation of the doping area mainly to the surface of the semiconductor.
To obtain materials with higher photocatalytic activity induced by visible light it is necessary to increase the amount of photoactive Rh species with simultaneous inhibition of recombination processes. The preparation method which can lead to growth of concentration of active and homogenously spread Rh3+ species able for reversible redox processes incorporated mainly into the surface of titania particles should cause improvement of the activity resulting from two-step bandgap excitation. Moreover, bringing doping area to the surface and subsurface of host material may reduce undesired consequence of bulk doping, such as accelerated recombination. Created Rh species, due to the creation of new electronic states within the bandgap resulting from the strong interaction with TiO2 lattice could be called built-in redox mediators.
Experimental
Materials and methods
Characterization of the materials
Acknowledgements
The research was supported by the Polish Ministry of Science and Higher Education within the Iuventus Plus grant (No. IP 2012030572) and Ideas Plus grant (0003/ID/2012/62). STEM/EDX measurements have been carried out with equipment purchased with financial support from the European Regional Develop-ment Fund in the framework of the Polish Innovation Economy Operational Program (Contract No. POIG.02.01.00-12-023/08).Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, Chem. Lett., 2003, 32, 772 CrossRef CAS.
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33 CrossRef CAS.
- J. Choi, H. Park and M. R. Hoffmann, J. Phys. Chem. C, 2010, 114, 783 CAS.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, J. Photochem. Photobiol., A, 2002, 148, 71 CrossRef CAS.
- Y. Sasaki, A. Iwase, H. Kato and A. Kudo, J. Catal., 2008, 259, 133 CrossRef CAS.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669 CrossRef.
- K. Wilke and H. D. Breuer, J. Photochem. Photobiol., A, 1999, 121, 49 CrossRef CAS.
- K. Song, X. Han and G. Shao, J. Alloys Compd., 2013, 551, 118 CrossRef CAS.
- Z. Y. Tan, L. L. Wang, Y. C. Yang and W. Z. Xiao, Eur. Phys. J. B, 2012, 85, 138 CrossRef.
- K. K. Ghuman and C. V. Singh, J. Phys.: Condens. Matter, 2013, 25, 1 Search PubMed.
- R. Niishiro, R. Konta, H. Kato, W.-J. Chun, K. Asakura and A. Kudo, J. Phys. Chem. C, 2007, 111, 17420 CAS.
- R. Niishiro, S. Tanaka and A. Kudo, Appl. Catal., B, 2014, 187–196 CrossRef CAS.
- R. Konta, T. Ishii, H. Kato and A. Kudo, J. Phys. Chem. B, 2004, 108, 8992 CrossRef CAS.
- F. E. Oropeza and R. G. Egdell, Chem. Phys. Lett., 2011, 515, 249 CrossRef CAS.
- Z.-M. Dai, G. Burgeth, F. Parrino and H. Kisch, J. Organomet. Chem., 2009, 694, 1049 CrossRef CAS.
- S. Kitano, K. Hashimoto and H. Kominami, Chem. Lett., 2010, 39, 627 CrossRef CAS.
- S. Kitano, N. Murakami, T. Ohno, Y. Mitani, Y. Nosaka, H. Asakura, K. Teramura, T. Tanaka, H. Tada, K. Hashimoto and H. Kominami, J. Phys. Chem. C, 2013, 117, 11008 CAS.
- K. Furuhashi, Q. Jia, A. Kudo and H. Onishi, J. Phys. Chem. C, 2013, 117, 19101 CAS.
- J. Z. Bloh, R. Dillert and D. W. Bahnemann, J. Phys. Chem. C, 2012, 116, 25558 CAS.
- J. Z. Bloh, R. Dillert and D. W. Bahnemann, ChemCatChem, 2013, 5, 774 CrossRef CAS.
- T. Ikeda, T. Nomoto, K. Eda, Y. Mizutani, H. Kato and A. H. O. Kudo, J. Phys. Chem. C, 2008, 112, 1167 CAS.
- J. Kuncewicz and B. Ohtani, Chem. Commun., 2015, 51, 298 RSC.
- M. Zimowska, J. B. Wagner, J. Dziedzic, J. Camra, B. Borzęcka-Prokop and M. Najbar, Chem. Phys. Lett., 2006, 417, 137 CrossRef CAS.
- Z. Weng-Sieh, R. Gronsky and A. T. Bell, J. Catal., 1997, 170, 62 CrossRef CAS.
- S. Suhonen, M. Valden, M. Hietikko, R. Laitinen, A. Savimaki and M. Harkonen, Appl. Catal., A, 2001, 218, 151 CrossRef CAS.
- K. Iwashina and A. Kudo, J. Am. Chem. Soc., 2011, 133, 13272 CrossRef CAS PubMed.
- S. Kawasaki, K. Akagi, K. Nakatsuji, S. Yamamoto, I. Matsuda, Y. Harada, J. Yoshinobu, F. Komori, R. Takahashi, M. Lippmaa, C. Sakai, H. Niwa, M. Oshima, K. Iwashina and A. Kudo, J. Phys. Chem. C, 2012, 116, 24445 CAS.
- E. Świętek, K. Pilarczyk, J. Derdzińska, K. Szaciłowski and W. Macyk, Phys. Chem. Chem. Phys., 2013, 15, 14256 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The spectra of the used light source; the changes in rutile lattice constant with Rh concentration; diffuse reflectance spectra of Rh2O3 and TiO2 mixed with Rh2O3; diffuse reflectance spectra of Rh2O3·mixed TiO2 and calcined irradiated with various light ranges; differential absorption spectra of 0.01% Rh–TiO2 irradiated with various light ranges in various conditions; double beam photocatalytic tests; the changes in the KM function value at 440 nm determined for not calcined samples containing various Rh concentrations; diffuse reflectance spectra and photocatalytic activity determined for rinsed 0.001% Rh–TiO2. See DOI: 10.1039/c6ra09364g |
| This journal is © The Royal Society of Chemistry 2016 |