
A novel sulfur-impregnated porous carbon matrix as a cathode material for a lithium–sulfur battery†
Yanhui Cuia,
Xiao Lianga,
Jue Ouyanga,
Jiayi Huanga,
Jiong Zenga,
Junwei Wu*a,
Zuohua Li*b,
Chenqiang Duc,
Zhoufu Lic,
Andrew P. Bakera,
Kevin Huangd and
Xinhe Zhange
aShenzhen Key Laboratory of Advanced Materials, Department of Materials Science and Engineering, Harbin Institute of Technology Shenzhen Graduate School, Shenzhen, 518055, China. E-mail: junwei.wu@hitsz.edu.cn; Fax: +86-755-2603-3504; Tel: +86-755-2603-3290
bSchool of Civil and Environmental Engineering, Harbin Institute of Technology Shenzhen Graduate School, Shenzhen 518055, China. E-mail: lizuohua@hitsz.edu.cn; Fax: +86-755-2603-3509; Tel: +86-755-2603-4949
cDepartment of Applied Chemistry, School of Chemical and Engineering, Tianjin University, Tianjin, 300072, China
dDepartment of Mechanical Engineering, University of South Carolina, Columbia, SC 29201, USA
eDongguan Mcnair Technology Co., LTD, Dongguan, 523800, China
First published on 29th June 2016
Abstract
A novel sulfur-impregnated porous carbon matrix (PCM-Z-S) has been prepared as a cathode material for a lithium–sulfur battery. The porous carbon matrix (PCM-Z), which was obtained using de-waxed cotton and ZnCl2 as an activator, has a surface area of 1056 m2 g−1 and a pore volume of 1.75 cm3 g−1. The PCM-Z was mixed with sublimed sulfur and then heated in nitrogen gas to form a carbon–sulfur 58 wt% composite (PCM-Z-S) which has excellent electrochemical proprieties. The PCM-Z-S delivers a capacity of 850 mA h g−1 at 1C and retains 630 mA h g−1 after nearly 200 cycles which are values much higher than that of a carbon matrix prepared without ZnCl2. These results show the sulfur-impregnated porous carbon matrix (PCM-Z-S) has great potential as a cathode material in a lithium–sulfur battery.
Introduction
The development of high capacity energy storage systems is very important for the development of portable electronic devices, power tools, and electric vehicles.1–4 Lithium–sulfur (Li–S) batteries have attracted more and more attention as potential energy storage devices because the sulfur cathode has both a high theoretical capacity of 1672 mA h g−1 and an energy density of 2600 W h kg−1, which are 3–5 times higher than conventional cathodes.5–10 There are three major issues that impede the practical applications for a sulfur cathode in Li–S batteries.3–5 Firstly, sulfur is insulating at room temperature which impedes any electrochemical reaction. Secondly, there is interconversion between cyclo-S8, soluble lithium polysulfides Li2Sx (3 ≤ x ≤ 8), and insoluble sulfides Li2S2/Li2S during charge–discharge process. These, along with a series of structural and morphological changes, lead to an unstable electrochemical contact within sulfur electrodes.4,5 Thirdly, the dissolved polysulfides shuttle between the anode and cathode during the charge–discharge process allowing side reactions that involve reduction with lithium anode and reoxidation reactions at the cathode which result in a low utilization of active material, poor cycle life, and low system efficiency.11 Various strategies have been developed to alleviate these issues including blocking lithium sulfide across a functional membrane,12–14 covering the lithium anode with inert materials to limit reaction with lithium sulfide,15,16 and restricting sulfur to escape from a conductive matrix.17–19 A carbon matrix allows conventional impregnation of sulfur and is effective in enhancing electrochemical performance. Consequently various carbon structures have been used such as carbon spheres with micropores,17 spherical ordered mesoporous carbon with a large number of inner pores,18 porous hollow carbon,19 graphene oxide,20,21 porous carbon nanofibers and hollow carbon nanofibers or nanotubes.22–24 However the complex preparation and, in some cases, the high cost of the above processes limit their commercialization.The biomass exists abundantly on the earth and can be used to prepare porous carbon easily and massively. If this kind of porous carbon can be used as host for sulfur, the cost of Li–S battery will be reduced. In this paper, de-waxed cotton was used as carbon source to prepare porous carbon matrix (PCM) because cotton can be planted abundantly on the earth and its attractive fiber morphology.25,26 Additionally, acidified zinc chloride (ZnCl2) can hydrolyze cellulose in a pretreatment stage and restrict formation of tars which can block pore channels of carbon matrix in the carbonization process.27,28 Therefore, ZnCl2 solution has been used as activator to prepare a porous carbon matrix (PCM-Z). For impregnating ZnCl2 completely, cotton was pre-treated long time because the amount and the distribution of ZnCl2 in the precursor govern the porosity of the product. The PCM-Z derived from de-waxed cotton using this method has a surface area of 1056 m2 g−1 and a pore volume of 1.75 cm3 g−1 which is larger than previous reports for PCMs.25,26 A composite of PCM-Z with 58 wt% sulfur (PCM-Z-S) shows excellent electrochemical proprieties in contrast to a composite made using a porous carbon matrix formed without the use of acidified ZnCl2 (PCM-0) and impregnated with 54 wt% sulphur (PCM-0-S).
Experimental
A quantity of de-waxed cotton (5 g) was immersed in 100 ml aqueous solution of 0.25 M HCl containing 33 wt% ZnCl2. The solution with cotton was placed in an ultrasonic bath for 30 min and stewed at 70 °C for 24 h to ensure adequate impregnation of ZnCl2. The excess liquid was removed and the beaker with cotton was placed in an oven at 100 °C for 20 min to evaporate of the remaining liquid and then cooled down to 65 °C and held at this temperature for 14 days. Over this time it was observed (see ESI data Fig. S1†) that after 3 days the cotton had dissolved forming a transparent brown solution and after 14 days the color had changed to black, suggesting the cotton had partially carbonized. The mixture was then heated for 30 min at 100 °C and 1 h at 550 °C under nitrogen atmosphere in furnace to further promote carbonization and then boiled for 3 h in 1 M HCl solution at 100 °C followed by washing with hot distilled water to remove any remaining ZnCl2. After drying for 4 h at 80 °C the product was ground by mortar and pestle and then thermally treated for 90 min at 1000 °C under nitrogen atmosphere to obtain the PCM-Z product. The preparation of PCM-0 is the same as that of PCM-Z but without ZnCl2 addition.The carbon–sulfur composites were prepared by mixing PCM-Z and PCM-0 with sublimed sulfur together in a mass ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5 and 1:2 respectively and then heating the mixture for 10 h at 155 °C under a nitrogen atmosphere.
:1.5 and 1:2 respectively and then heating the mixture for 10 h at 155 °C under a nitrogen atmosphere.
Characterizations
The morphology and composition for PCM-0, PCM-Z, and the sulfur composites were examined by scanning electron microscopy (SEM) (Hitachi S4700). A thin Au layer was coated on all the samples to improve the image quality. N2 adsorption isotherms were obtained by using a Belsorp (Japan) to investigate the external and specific surface area. The PCM-0, PCM-Z and their sulfur composites were preheated above 100 °C for 3 h at 0.1 kPa prior to measurement. The sulfur content in the PCM-0-S and PCM-Z-S composites was measured in a NETZSCH STA 449F3 thermal analyzer in a N2 atmosphere at a heating rate of 10 °C min−1.Electrochemical measurements
Electrochemical tests were performed with CR2032 coin type cells. The cathodes were prepared from a mixture of the PCM-0-S or PCM-Z-S composites, super P, and PVDF in N-methyl-2-pyrrolidone, with a weight ratio of 80:10:10. The obtained slurry was pasted onto aluminium foil with a uniform thickness of 200 μm, and dried at 50 °C for 24 h. Lithium metal was used as the anode and reference electrode and the electrolyte was 1.0 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) and 0.1 M LiNO3 in a mixture of 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME) (1:1, v/v). The coin cells were assembled in an argon-filled glove box (Mikrouna Universal 2440/750). Galvanostatic charge–discharge tests were performed in the range 1.7–2.6 V at different rates by using a LAND-CT2001A battery testing instrument. Cyclic voltammograms (CVs) were obtained at a scan rate of 0.1 mV s−1 in the range 1.7–2.6 V with an electrochemical workstation (CHI 1040, CH instrument, China). The electrochemical measurements were all conducted at room temperature.
Results and discussion
XRD patterns, Raman spectra, surface areas and morphology of PCM-0, PCM-Z, PCM-0-S and PCM-Z-S
The XRD patterns of sublimed sulfur used for PCM impregnation, PCM-Z, PCM-0, and the PCM-0-S and PCM-Z-S composites are shown in Fig. 1(a). The XRD pattern obtained for sublimed sulfur can be assigned to pure crystalline sulfur (PDF#08-0247, cyan line in Fig. 1(a)). The XRD patterns for PCM-0 and PCM-Z show two broad diffraction peaks located at about 22.5° and 44° 2 theta which correspond to the (002) and (100) planes, respectively. The (002) peak ∼22.5° indicates the presence of partially graphitized structures in the PCM and the (100) peak ∼44° represents amorphous frameworks.29 The XRD pattern of PCM-Z shows more background noise than that of PCM-0 which indicates a less crystalline structure. For further comparing the proportion of graphitized and amorphous structures, Raman spectroscopy was used and the results are shown in Fig. 1(b). The two peaks around 1583 and 1343 cm−1 correspond to the well-known G and D bands of carbon. The D band and G band are associated with the disordered and graphitized structure respectively. The ratio of the D to G band intensity (ID/IG) is commonly used to evaluate the crystallinity degree of the carbon materials.30 The ID/IG ratios are 1.5 and 1.46 for PCM-Z and PCM-0 respectively indicating a more amorphous structure for PCM-Z which agrees with their respective XRD patterns. The XRD pattern of PCM-Z-S composite in Fig. 1(a) shows no obvious diffraction peaks that belong to sublimed sulfur (see XRD pattern in green Fig. 1(a)) suggesting sulfur is impregnated into the pores of PMC-Z and exists in an amorphous form. As shown by EDS analysis (see ESI data in Fig. S2†) the sulfur distribution is uniform throughout the composite. The XRD pattern of the PCM-0-S composite shows obvious diffraction peaks similar to that of sublimed sulfur suggesting sulfur is not fully impregnated into the pores of PMC-0 and exists mainly in a crystalline form. In the case of PCM-Z-S (Fig. 1(a) XRD pattern shown in pink) there is an increase in the intensity of the amorphous hump ∼22.5° 2 theta which coincides with the 2 theta values of the main diffraction peaks for sublimed sulfur. Further evidence of the sulfur impregnation in PCM-Z-S is given by thermal analysis (TGA curves shown in ESI data Fig. S4†), which shows a higher sulfur loss from PCM-Z-S (58 wt%) compared to PCM-0-S (54 wt%) and a higher temperature required to remove most of the sulfur from PCM-Z-S compared to PCM-0-S due to the sulfur being impregnated into the pores. | ||
| Fig. 1 (a) XRD patterns of PDF#08-0247, sulfur, PCM-0, PCM-Z, PCM-0-S and PCM-Z-S. (b) Raman spectra of PCM-0 and PCM-Z. (c) N2 adsorption and desorption curves of PCM-0 and PCM-Z. (d) Pore radius distribution of the PCM-0 and PCM-Z. | ||
This can be attributed to the lower surface area and smaller pore volume of PCM-0 shown by the BET analysis for the surface area in Fig. 1(c) and pore volume 1(d). The N2 adsorption and desorption curves in Fig. 1(c) for PCM-0 and PCM-Z show that PCM-Z falls into the category of type IV, i.e. mesoporous materials31,32 in contrast to that of PCM-0. The surface area and pore volume of PCM-Z are 1056 m2 g−1 and 1.75 cm3 g−1 respectively but the surface area and pore volume of PCM-0 are 448 m2 g−1 and 0.026 cm3 g−1 respectively (see ESI data Table S1†). As seen in Fig. 1(d) the pore radii of PCM-Z are distributed below 15 nm and for PCM-0 below 2.5 nm which confirms ZnCl2 gives PCM-Z a larger surface area and pore volume allowing for more effective sulfur impregnation.
The SEM images of PCM-0, PCM-0-S, PCM-Z and PCM-Z-S are shown in Fig. 2. The SEM image of PCM-0 in Fig. 2(a) shows a smooth surface morphology which is expected from the small pore size (<2.5 nm) but the morphology of the PCM-0-S surface appears rough; which can be attributed to crystalline sulphur, as shown by the XRD pattern in Fig. 1(a), on the surface of PCM-0. In contrast to PCM-0, the surface morphology of PCM-Z shown in Fig. 2(c), is porous and when impregnated with sulfur the morphology of PCM-Z-S changes little even though the sulfur content is 4% higher than PCM-0-S. This can be explained by the pore volume decreasing drastically from 1056 cm2 g−1 for PCM-Z to 153 cm2 g−1 for PCM-Z-S (see ESI data Table S1†) which is due to sulfur impregnating PCM-Z. Confirmation of this is shown by high resolution images (see ESI data Fig. S6(a) and S6(b)† for PCM-Z and PCM-Z-S respectively) where lattice spacings of 0.473 nm are observed for PCM-Z-S that correspond to (202) lattice plane of sulfur (PDF#08-0247) along with blurred lattice fringes indicating poor crystallization of sulfur which is in agreement with the XRD results in Fig. 1(a).
 | ||
| Fig. 2 SEM images of (a) PCM-0, (b) PCM-0-S, (c) PCM-Z and (d) PCM-Z-S. All scale bars = 1.00 micron. | ||
Electrochemical performances
The electrochemical performance of PCM-0-S and PCM-Z-S composites is shown in Fig. 3. The CV plot for PCM-0-S in 3(a) and for PCM-Z-S 3(b) show that from high to low voltage the reduction curves has two obvious cathodic reduction peaks which can be attributed to the reduction of the cyclo-S8 to Li2Sn (6 ≤ n ≤ 8) and Li2Sn (6 ≤ n ≤ 8) to Li2Sm (2 ≤ m ≤ 4).4,5 While from low to high voltage the cathodic oxidation curves also show two oxidation peaks which can be attributed to the oxidation of Li2S to polysulfide and then to cyclo-S8. In the case of PCM-0-S the first reduction peak located at 2.28 V shifts to the right and the current increases with cycling which indicates an activation process (the forth cycle curve is close to the fifth and omitted for conciseness). Also the area of the second reduction peak is smaller than that of the first reduction peak indicating an incomplete reduction to Li2Sm (2 ≤ m ≤ 4) due to the small surface area and pore volume of PCM-0 (see BET results in Fig. 1(c) and ESI data Table S1†). The same indication is given by the corresponding area of the oxidation peaks. While PCM-Z-S, along with cycling, shows the cathodic reduction peaks also shifting to the right and becoming sharper which indicates an activation process due to the electrolyte being impeded in accessing the internal surface33,34 of the PCM-Z. The electrochemical impedance spectrums for the PCM-0-S (54 wt% sulfur) and PCM-Z-S (58 wt% sulfur) composites are shown in Fig. 3(c). Each spectrum includes a semicircle in high to medium frequency region and an inclined line in the lowest frequency region. The high to medium frequency semicircle is attributed to the charge transfer resistance (Rct) and the low-frequency sloped line corresponds to ion diffusion within the cathodes.35 In the equivalent circuit inset in Fig. 3(c), Re is the resistance of electrolyte, Rct/CPE2 is the charge transfer resistance and its relative capacitance. The Rct of PCM-Z-S is smaller than PCM-0-S because the large surface area and pore volume of PCM-Z allows better contact between sulfur and the carbon matrix.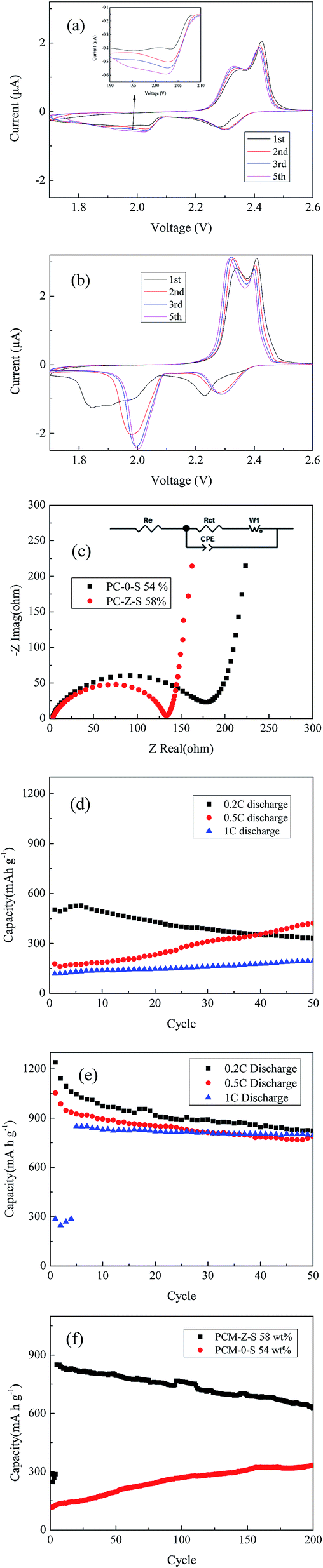 | ||
| Fig. 3 (a) Typical cyclic voltammograms of PCM-0-S 54 wt% composite and (b) typical cyclic voltammograms of PCM-Z-S 58 wt% composite, (c) electrochemical impedance spectrum of PCM-0-S 54 wt% and PCM-Z-S 58 wt% composite electrodes in Li–S batteries before initial discharge, (d) the cycling performance of PCM-0-S 54 wt% composite at different C values (e) the cycling performance PCM-Z-S 58 wt% composite at different C values and (f) long cycling performance of PCM-0-S 54 wt% composite and PCM-Z-S 58 wt% composite at 1C. | ||
The cycling performance of PCM-0-S and PCM-Z-S composites at 0.2C, 0.5C and 1C (the current at 1C is equivalent to 1672 mA g−1 based on sulfur) are shown in Fig. 3(d) and (e). The capacity at 0.2C initially increases for PCM-0-S, shown in Fig. 3(d), and then decreases indicating an activation process due to the small surface area and pore volume. As the current increases (0.5C and 1C) the activation process takes longer because the larger currents result in larger polarization leading to incomplete charge/discharge. The capacities for PCM-Z-S are much higher, as shown in Fig. 3(e), than obtained for PCM-0-S at 0.2C, 0.5C, and 1C because of its larger surface area and pore volume. However at 1C, the capacity shows a drastic increase after first several cycles whose second discharge plateaus disappear (see ESI data Fig. S3(a)†). While Fig. S5† shows the voltage of second discharge plateau decreases with increasing applied current because large current results in large polarization30,36 possibly due to of sulfur aggregating on a micron scale. The supersaturation point of lithium polysulfides in the discharge process7,36 also becomes obvious at large current (see Fig. 3(b)). Once the supersaturation point voltage drops below 1.7 V, deep discharge cannot proceed further and only low capacity can be delivered due to above reasons (see ESI data Fig. S3(a)†). However along with the activation process, the aggregated sulfur will redeposit and become uniform35 which allows the electrolyte to gradually become in contact with sulfur deeply impregnated in the PCM-Z which then becomes electrochemically active resulting in the corresponding gradual increase in capacity. As more sulfur becomes electrochemically active the supersaturation and polarization decrease leading to the increase of the second discharge plateau. Once the quantity of active sulfur reaches the threshold (the second discharge plateau increases above 1.7 V), the discharge can proceed and there is an increase in the capacity of the fifth cycle. It should be noted that, with longer deposition time the activation process does not disappear but can be alleviated (see ESI data Fig. S3(b)†).
Regardless of the activation process, the PCM-Z-S composite obtains first discharge capacities of 1240 and 1053 mA h g−1 at 0.2C and 0.5C, respectively. And at the fiftieth discharge the capacities are 823 and 787 mA h g−1, showing a decrease of 33.6% and 25.4%, respectively. The ratio of the decrease at large current is smaller than that at lower current especially at 1C current where the large capacity and the fiftieth capacity are 850 mA h g−1 and 794 mA h g−1 respectively, showing a decrease of only 6.6%. The reason for this can be the higher C values shorten the escape time of soluble lithium polysulfides per cycle which leads to good cycling performance. The cycling performance of the PCM-0-S and PCM-Z-S composites is shown in Fig. 3(f). The capacity of PCM-S 58 wt% composite increases to 850 mA h g−1 at 1C and retains 630 mA h g−1 after nearly 200 cycles, showing a fading of 0.13% per cycle. Although the activation process of PCM-0-S 54 wt% composite at 1C is continuous over 200 cycles the capacity is very much lower than the PCM-S 58 wt% composite.
Conclusions
Large surface area and pore volume PCM-Z has been prepared from cotton using acidified ZnCl2 solution for activation. When PCM-Z is formed into a composite with sulfur (PCM-Z-S 58 wt%), and used in a Li–S battery as a cathode, a high capacity of 850 mA h g−1 was obtained after several cycles at 1C and 630 mA h g−1 after 200 cycles. The excellent battery performance using the PCM-Z-S 58 wt% composite suggests this sulfur-impregnated porous carbon matrix is suitable for application in a lithium–sulfur battery. Future work such as surface modification will be investigated to further improve the performance of carbon–sulfur composites as a cathode material for a lithium–sulfur battery.Acknowledgements
This work is supported by the International Collaboration Program of Shenzhen (GJHZ20150312114008636, GJHZ201304 11141529167), Shenzhen Peacock Plan Program (KQCX201 40521144358003), Fundamental Research Program of Shenzhen (JCYJ20140417172417144, JCYJ20130329141705731), Shenzhen Technology Innovation Program-Technology Development Projects (Grant No. CXZZ20140904154839135), Shenzhen Personal Maker Program (GRCK2015092816025894), and Guangdong Innovative and Entrepreneurial Research Team Program (No. 2013N079).Notes and references
- A. Manthiram, Y. Fu, S. H. Chung, C. Zhu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018–3032 RSC.
- A. Manthiram, Y. Fu and Y. S. Su, Acc. Chem. Res., 2012, 46, 1125–1134 CrossRef PubMed.
- Y. Son, J. S. Lee, Y. Son, J. H. Jang and J. Cho, Adv. Energy Mater., 2015, 5, 1500110 Search PubMed.
- L. F. Nazar, M. Cuisinier and Q. Pang, MRS Bull., 2014, 39, 436–442 CrossRef CAS.
- S. Zhang, K. Ueno, K. Dokko and M. Watanabe, Adv. Energy Mater., 2015, 5, 1500117 Search PubMed.
- X. Ji and L. F. Nazar, J. Mater. Chem., 2010, 20, 9821–9826 RSC.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2012, 46, 1135–1143 CrossRef PubMed.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS.
- Y. Chen, N. Liu, H. Shao, W. Wang, M. Gao, C. Li, H. Zhang, A. Wang and Y. Huang, J. Mater. Chem. A, 2015, 3, 15235–15240 CAS.
- J. Q. Huang, Q. Zhang, H. J. Peng, X. Y. Liu, W. Z. Qian and F. Wei, Energy Environ. Sci., 2014, 7, 347–353 CAS.
- Z. Liu, X. H. Zhang and C. S. Lee, J. Mater. Chem. A, 2014, 2, 5602–5605 CAS.
- C. Huang, J. Xiao, Y. Shao, J. Zheng, W. D. Bennett, D. Lu, L. V. Saraf, M. Engelhard, L. Ji, J. Zhang, X. Li, G. L. Graff and J. Liu, Nat. Commun., 2014, 5, 4015 Search PubMed.
- W. Li, J. Hicks-Garner, J. Wang, J. Liu, A. F. Gross, E. Sherman, J. Graetz, J. J. Vajo and P. Liu, Chem. Mater., 2014, 26, 3403–3410 CrossRef CAS.
- B. Zhang, X. Qin, G. R. Li and X. P. Gao, Energy Environ. Sci., 2010, 3, 1531–1537 CAS.
- J. Schuster, G. He, B. Mandlmeier, T. Yim, K. T. Lee, T. Bein and L. F. Nazar, Angew. Chem., Int. Ed., 2012, 51, 3591–3595 CrossRef CAS PubMed.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., Int. Ed., 2011, 123, 6026–6030 CrossRef.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS PubMed.
- N. Li, M. Zheng, H. Lu, Z. Hu, C. Shen, X. Chang, G. Ji, J. Cao and Y. Shi, Chem. Commun., 2012, 48, 4106–4108 RSC.
- L. W. Ji, M. M. Rao, S. Aloni, L. Wang, E. J. Cairns and Y. G. Zhang, Energy Environ. Sci., 2011, 4, 5053–5059 CAS.
- G. Zheng, Y. Yang, J. J. Cha, S. S. Hong and Y. Cui, Nano Lett., 2011, 11, 4462–4467 CrossRef CAS PubMed.
- J. Guo, Y. Xu and C. Wang, Nano Lett., 2011, 11, 4288–4294 CrossRef CAS PubMed.
- S. Wang, Z. Ren, J. Li, Y. Ren, L. Zhao and J. Yu, RSC Adv., 2014, 4, 31300–31307 RSC.
- G. Ma, D. Guo, K. Sun, H. Peng, Q. Yang, X. Zhou, X. Zhao and Z. Lei, RSC Adv., 2015, 5, 64704–64710 RSC.
- M. Kitano, K. Arai, A. Kodama, T. Kousaka, K. Nakajima, S. Hayashi and M. Hara, Catal. Lett., 2009, 131, 242–249 CrossRef CAS.
- F. Schüth, K. S. W. Sing and J. Weitkamp, Handbook of porous solids, Wiley-Vch, 2002, vol. 3, pp. 1785–1795 Search PubMed.
- S. Wei, H. Zhang, Y. Huang, W. Wang, Y. Xia and Z. Yu, Energy Environ. Sci., 2011, 4, 736–740 CAS.
- K. Zhang, Q. Zhao, Z. Tao and J. Chen, Nano Res., 2013, 6, 38–46 CrossRef CAS.
- Y. S. Su and A. Manthiram, Nat. Commun., 2012, 3, 1166 CrossRef PubMed.
- Z. Fan, Y. Liu, J. Yan, G. Ning, Q. Wang, T. Wei, L. Zhi and F. Wei, Adv. Energy Mater., 2012, 2, 419–424 CrossRef CAS.
- L. Xiao, Y. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS PubMed.
- Y. Cui, J. Chen, K. Huang, C. Du, J. Wu, A. P. Baker and X. Zhang, RSC Adv., 2016, 6, 9117–9123 RSC.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. She and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef CAS PubMed.
- J. R. Akridge, Y. V. Mikhaylik and N. White, Solid State Ionics, 2004, 1, 243–245 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: (S1) The pre-carbonized process of PCM-Z, (S2) the SEM image of PCM-Z-S 58 wt% and EDX element mapping of S, (S3) the discharge–charge curves of PCM-S 58 wt% composite at 1C after deposited 1 and 8 days, (S4) the Tg analysis of PCM-0-S and PCM-Z-S, (S5) the first discharge and charge curves, of PCM-Z-S at 0.2C and 0.5C, (S6) high resolution images of PCM-Z and PCM-Z-S, (S7) calculated total surface area, total pore volume, and average pore size for PCM-0, PCM-0-S, PCM-Z and PCM-Z-S. See DOI: 10.1039/c6ra09338h |
| This journal is © The Royal Society of Chemistry 2016 |