
A simple regenerable electrochemical aptasensor for the parallel and continuous detection of biomarkers
Chunyan Liu,
Xi Liu,
Yun Qin,
Chunyan Deng* and
Juan Xiang
College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, P. R. China. E-mail: dengchunyan@csu.edu.cn; Fax: +86-731-88879616; Tel: +86-731-88876490
First published on 2nd June 2016
Abstract
In this present work, a simple regenerable electrochemical aptasensor for the parallel and continuous detection of protein biomarkers is reported. Mucin 1 protein (MUC1) and cinoembryonic antigen (CEA) were employed as the models of protein biomarkers. The sensing interface was modified by MUC1 aptamer and methylene blue (MB)-modified CEA aptamer via hybridization with a linker DNA. Herein, MB served as a signal transducer, MUC1 and CEA were measured in parallel, and the detection limits of MUC1 and CEA were 0.13 nM and 2.75 ng mL−1, respectively. More significantly, the present biosensing interface can also be employed for the continuous detection of biomarkers. After the detection of MUC1, the resulting biosensing interface can be continuously used to measured CEA, and the detection limit was obtained as 0.5 ng mL−1, which was obviously lower than that based on the parallel analysis of CEA. Meanwhile, the aptasensor can be regenerated by rehybridization of DNA2 or DNA3-MB after the parallel detection of targets, or by introducing MUC1 and CEA and then rehybridizing the MUC1 aptamer and CEA aptamer with linker DNA1 on the gold electrode. Therefore, it can be elucidated that this present strategy provides different protocols (parallel and continuous detection) to meet the requirements of analytical applications. This present method is also cost-effective and operationally simple compared with instrument-based methods for the multiplexed detection of markers. And the accuracy, simplicity and reproducibility of this biosensor are excellent. It will give potential guidance for the multiplexed detection of biomarkers or other small molecules.
1. Introduction
It has been widely reported that protein biomarkers can be used as indicators of different biological processes, including normal processes, pathogenic processes, or pharmacological responses.1–4 Therefore, it is important to carry out the detection of protein biomarkers to realize disease recognition. Currently, it is well known that specific protein recognition ligands, especially antibodies, have long been used for protein biomarkers because antibodies are of high specificity and high binding affinities to many targets.5–7 And therefore, by combing with various analytical techniques, such as fluorescence, chemiluminescence, and electrochemistry, a great many immunoassay biosensors and diagnostic tools have been developed.8–12 However, in case of antibodies, there are some disadvantages such as complexity in production, high cost and poor instability.13 Accordingly, it is essential to pursue novel protein recognition ligands for the detection of biomarkers.Aptamers as artificial oligonucleotides are selected through systematic evolution of ligands by exponential enrichment (SELEX). They can specifically bind many targets, such as small molecules, proteins, and even intact viruses and cells.14–16 Aptamers have been widely employed for molecular analyses in research and medical diagnostics because they possess various advantages over antibodies, such as low molecular weight, excellent stability, easy synthesis, programmable base sequences and structures.17–19 By using different measuring techniques, a variety of aptasensors have been developed. For example, Sun et al. carried out the highly efficient colorimetric detection of ATP utilizing a split aptamer target binding strategy and superior catalytic activity of graphene oxide–platinum/gold nanoparticles,20 Pu et al. reported an electrochemical strategy with molecular beacon and hemin/G-quadruplex for the detection of Clostridium perfringens DNA on screen-printed electrodes.21 Zhu group developed a binding-induced fluorescence turn-on assay using aptamer-functionalized silver nano cluster DNA probes.22 In these aptamer-based detection strategies, the one designed biosensor can be used to detect only one protein biomarker.
On the other hand, it is well known that for a certain disease, there usually are the abnormal changes of several protein biomarkers, the detection of only one protein biomarker may result in the false positive diagnosis of disease. Therefore, it is crucial to explore a kind of biosensor which can effectively and accurately measure two or more protein biomarkers, and it would further promote the practical application of these biosensor. In recent years, more time and energy has been devoted to these related studies by researchers. By taking advantage of the unique properties of analytical techniques, such as microfluidic,23 atomic force microscopy (AFM),24 and ISET-MS,25 the multiplexed analysis of targets has been realized, but expensive instruments and skilled operators were required. In addition, some multifunctional electrochemical aptasensors have been explored for the parallel analysis of targets. For example, Wang et al., has reported the label-free bifunctional electrochemiluminescence aptasensor for the parallel detection of adenosine and lysozyme based on switching structures of aptamers from DNA/DNA duplex to DNA/target complex.26 Du et al. fabricated a multifunctional label-free electrochemical biosensor based on an integrated aptamer for parallel detection of adenosine triphosphate (ATP) and thrombin.27 It also has been reported that a sensitive bifunctional aptamer-based electrochemical biosensor for small molecule and proteins was simply fabricated based on the AuNPs signal amplification.28 Using these biosensors, the parallel detection of two or more molecules can be realized, but it should be paid attention to that the multiplexed detection could not be carried out by using the same biosensing interface. That is, each biosensing interface can be used only one time for the detection of one target. This may cause the decrease in the reproducibility of biosensors, and make the detection more complex. Therefore, it can be elucidated that there is significant challenge for the multiplexed detection in the practical application. Especially reproducibility and simplicity of these reported biosensors still need to be further improved, which are always neglected.
In this paper, a simple regenerable electrochemical aptasensor for the parallel and multiplexed detection of biomarkers was fabricated. Herein, two protein biomarkers, mucin 1 protein (MUC1) and cinoembryonic antigen (CEA) were used as models, respectively. MUC1 as a well-known tumor marker, has excessive expression in a variety of malignant tumors, such as breast cancer, stomach cancer, colorectal cancer, lung cancer, prostate, ovarian, bladder cancer. CEA, a tumor marker of glycoprotein, widely distributes in many tumor cells. Therefore, the direct sensitive measurement of MUC1 and CEA can be significant for the early diagnosis, differentiating diagnosis, curative effect monitoring, and follow-up examinations of patients with tumors or carcinomas. Correspondingly, two DNA strands (MUC1 aptamer and MB modified CEA aptamer) were immobilized on the gold electrode by hybridization with the linker DNA (DNA1) to form the biosensing interface. The methylene blue (MB) serves as a signal transducer for the detection of biomarkers. Using this strategy, MUC1 and CEA can be parallel analyzed. More significantly, MUC1 and CEA can be continuously measured just using the same biosensing interface. After the detection of MUC1, the obtained interface can be continuously used to detect CEA with higher sensitivity. And moreover, after the parallel detection of MUC1, CEA or the continuous detection of MUC1 and CEA, the biosensing interfacing can be regenerated by hybridization of DNA2, DNA3-MB or DNA2 and DNA3-MB, which may be significant for improve the reproducibility and simplicity of the biosensor. This designed strategy may provide a method for the more accurate, simple and efficient multiplexed detection of targets, which provides a potential foundation for the development of aptamer-based biosensor in practical application, and it is also signification for the development of the electrochemical aptasensor for sensing other tumor markers. Furthermore, it will have important applications in the sensitive and selective electrochemical determination of other small molecules and proteins.
2. Experimental
2.1. Materials
6-Mercaptohexanol (MCH), tris-(hydroxymethyl)aminomethane (Tris), were purchased from Bio Basic Inc. Tumor necrosis factor-α (TNF-α), thrombin, NaCl, MgCl2·6H2O were obtained from Amersco. All oligonucleotides were purchased from Sangon Biotechnology Co., Ltd. (Shanghai, China), and sequences of all oligonucleotides were listed as follows: thiolated DNA1 (5′-HS-TTT TTT TAT CCA AAG TTT TTT CGA ATA AGT-3′), DNA2 (5′-GCA GTT GAT CCT TTG GAT ACC CTG G-3′) and MB modified DNA3 (5′-TTA ACT TAT TCG ACC ATA-MB-3′). DNA1 contains complementary sequence (bold) to DNA2 (bold). And also the sequence (italic) in DNA1 is complementary to DNA3 sequence (italic). DNA1, DNA2 and DNA3 were diluted to 10 μM in 34 mM Tris–HCl buffer (pH 7.4, 233 mM NaCl, 8.5 mM KCl, 1.7 mM MgCl2, 1.7 mM CaCl2) for use. The obtained solutions were stored at 4 °C in refrigerator before use. MUC1 and CEA were purchased from Shanghai Apeptide Co., Ltd. The peptides were suspended in 10 mM phosphate buffer solution (PBS, pH 7.4) to obtain different concentrations for the subsequent experiments. All other reagents are of analytical purity and used as received. All solutions were prepared using ultrapure water obtained from a Milli-Q water purification system (Millipore Corp., Billerica, MA).2.2. Preparation of the modified electrode
Prior to aptamer immobilization, the bare gold electrode (2 mm in diameter) was polished sequentially with 0.3 μm and 0.05 μm alumina slurry, followed by ultrasonic cleaning in ethanol and ultrapure water. Subsequently, the gold electrode was cleaned in piranha solution (v/v 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2SO4/H2O2) (Caution: piranha solution reacts violently with many organic materials and should be handled with great care), followed by treatment with nitric acid. Afterwards, the gold electrode was washed thoroughly with ultrapure water and dried under nitrogen gas. In order to modify the electrode, 20 μL of DNA1 solution was dropped onto the cleaned electrode and incubated for 16 h to form the self-assembled monolayer. After that, the gold electrodes were incubated in 1 mM 6-mercaptohexanol (MCH) solution for 1 h to block the unmodified region of the gold surface. Then a 20 μL DNA2 solution was covered onto the resulted Au/DNA1/MCH electrode and kept for 2 h to obtain the Au/DNA1/MCH/DNA2 electrode. Next a 20 μL DNA3-MB solution was dropped onto the electrode and kept for 2 h to formed the Au/DNA1/MCH/DNA2/DNA3-MB electrode. After each step, the electrodes were rinsed thoroughly with ultrapure water and dried under highly pure nitrogen stream prior to electrochemical characterization. For each step of immobilization, the electrochemical impedance measurements were performed in the presence of equimolar [Fe(CN)6]3−/4− as redox probe.
:1 H2SO4/H2O2) (Caution: piranha solution reacts violently with many organic materials and should be handled with great care), followed by treatment with nitric acid. Afterwards, the gold electrode was washed thoroughly with ultrapure water and dried under nitrogen gas. In order to modify the electrode, 20 μL of DNA1 solution was dropped onto the cleaned electrode and incubated for 16 h to form the self-assembled monolayer. After that, the gold electrodes were incubated in 1 mM 6-mercaptohexanol (MCH) solution for 1 h to block the unmodified region of the gold surface. Then a 20 μL DNA2 solution was covered onto the resulted Au/DNA1/MCH electrode and kept for 2 h to obtain the Au/DNA1/MCH/DNA2 electrode. Next a 20 μL DNA3-MB solution was dropped onto the electrode and kept for 2 h to formed the Au/DNA1/MCH/DNA2/DNA3-MB electrode. After each step, the electrodes were rinsed thoroughly with ultrapure water and dried under highly pure nitrogen stream prior to electrochemical characterization. For each step of immobilization, the electrochemical impedance measurements were performed in the presence of equimolar [Fe(CN)6]3−/4− as redox probe.
2.3. Apparatus and electrochemical measurements
Electrochemical impedance spectroscopy (EIS) measurements, and square wave voltammetry (SWV) were performed on a CHI650D electrochemical workstation. A conventional three-electrode system was used with the bare gold electrode as the working electrode, a platinum wire as counter electrode and a saturated calomel electrode (SCE) as reference electrode. All the potentials in this paper were in respect to SCE. All experiments were carried out at laboratory temperature.For the detection of protein biomarkers, the biosensor (Au/DNA1/MCH/DAN2/DNA3-MB electrode) was incubated in MUC1 or CEA solution (10 mM PBS, pH 7.4) with different concentrations at 37 °C for 2 h, followed by thoroughly washed with buffer to remove nonspecifically bound MUC1 or CEA. Then the electrochemical performance of the electrode was investigated by SWV with the potential step of 4 mV, the amplitude of 25 mV and the frequency of 15 Hz by scanning the potential from −0.40 to −0.15 V in PBS (10 mM, pH 7.4). Each measurement was repeated at least three times, and the control experiments for TNF-α and thrombin were carried out under the same conditions.
After the parallel detection of MUC1, CEA or the continuous detection of MUC1 and CEA, the biosensing interfacing can be regenerated by coating 20 μL DNA2, DNA3-MB, or 20 μL DNA2 and DNA3-MB onto the electrode surface for 2 h. After washing with 10 mM PBS, the regeneration of the aptasensor was realized. And the regenerated interface can be reused for another assay.
3. Results and discussion
3.1. Design strategy of the aptasensor
Scheme 1 shows the mechanisms for the parallel analysis of two protein biomarkers, and the continuous detection of two protein biomarkers using the same electrochemical sensing interface, respectively. Herein, MUC1 and CEA were used as the models of protein biomarkers, DNA1, DNA2, and the DNA3-MB were employed. The surface of the gold electrode was first self-assembled with the thiolated DNA1. The sulfhydryl-modified DNA1 as linker DNA was designed to be complementary with the DNA2 (MUC1 aptamer) and DNA3-MB (CEA aptamer). Therefore, DNA2 and DNA3-MB can be immobilized onto the gold electrode by hybridization with the linker DNA (DNA1) to form the biosensing interface (the Au/DNA1/MCH/DNA2/DNA3-MB electrode). Herein, MB on the biosensing interface (the Au/DNA1/MCH/DNA2/DNA3-MB electrode) was kept relatively far from the electrode surface and the electron transfer was essentially retarded. MB was used as the electro-active molecules to produce electrochemical signals for the detection of MUC1 and CEA.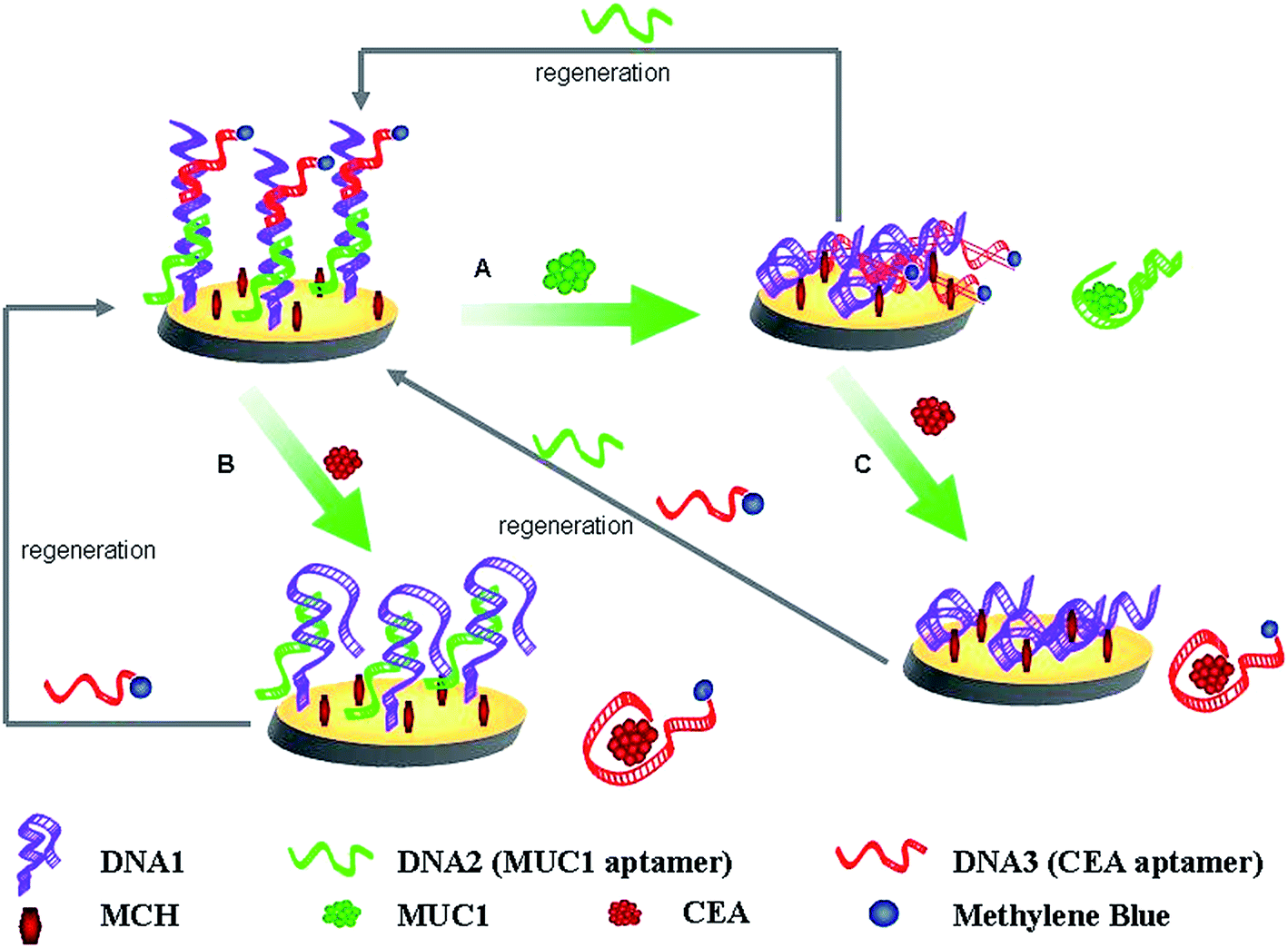 | ||
| Scheme 1 Schematic routines for the parallel or the continuous detection of MUC1 and CEA, and the regeneration of this present aptasensor. | ||
Just as shown in Scheme 1, MUC1 and CEA can be parallel analyzed. Upon MUC1 binding (Scheme 1A), the MUC1–aptamer complex dissociated from the modified electrode, and moved into solution. This resulted in that the distance between the redox moiety and the electrode surface was shortened, the electron transfer of MB was facilitated. And thus, the detection of MUC1 was realized by monitoring the MB signal enhancement. In addition, the biosensing interface can also be employed for the detection of CEA, just as shown in Scheme 1B. When the target CEA was introduced, the DNA3-MB/CEA complex divorced from the ds-DNA and MB signal was consequently reduced.
More importantly, the fabricated biosensing interface can be employed for the continuous detection of two protein biomarkers. After the fabricated interface was used to detect the concentration of MUC1, the current response of MB on the obtained electrode enhanced. And the resulted interface was continuously used to bind CEA (Scheme 1C), the CEA–aptamer complex dissociate from the modified electrode, which will lead to the more obvious current change of MB comparing to that based on the parallel detection of CEA (Scheme 1B).
On the other hand, it should be paid attention to that the biosensing interface can be simply regenerated after the parallel detection of MUC1, CEA, or the continuous detection of MUC1 and CEA by coating rehybridizing with DNA2, DNA3-MB, or DNA2 and DNA3-MB onto the electrode surface.
A simple and regenerable aptamer-based biosensor were fabricated based on the structure-switching properties of aptamers binding to their target molecules, which is generally applicable to many other aptamers.
3.2. Characterization of the modified electrode
The electrochemical properties of the modified electrodes were characterized by electrochemical impedance spectroscopy (EIS). According to literatures,29–31 the increase of the diameter of the semicircle reflects the increase of the interfacial charge-transfer resistance (Ret). On basis of the charge transfer kinetics of the [Fe(CN)6]3−/4− redox probe, faradaic impedance spectra with an [Fe(CN)6]3−/4− redox probe were modeled using the equivalent circuit approach. Fig. 1 displays the electrochemical impedance results of (a) the bare Au, (b) Au/DNA1, (c) Au/DNA1/MCH, (d) Au/DNA1/MCH/DNA2 and (e) Au/DNA1/MCH/DNA2/DNA3-MB electrodes. It is noted that the bare gold electrode shows a very small semicircle domain (Ret = 200 Ω, curve a), indicating a very fast electron transfer process of [Fe(CN)6]3−/4−.32 When DNA1 was self-assembled on the Au electrode surface, the charge-transfer resistance Ret enhanced to ca. 6000 Ω (curve b), indicating the electron transfer from [Fe(CN)6]3−/4− was hindered resulted from the negatively charged DNA1 repelling the [Fe(CN)6]3−/4− anions. Subsequent surface blocking with MCH also leaded to a significant increase in Ret (curve c in Fig. 1). After hybridization with DNA2 and DNA3-MB, Ret further increased to ca. 16000 Ω for DNA2 (curve d), and 21000 Ω for DNA3-MB (curve e), which was due to the negatively charged phosphate backbone of DNA repelling [Fe(CN)6]3−/4− anions from the electrode surface. All these experimental results proved that aptamers have been successfully immobilized on the gold electrode and the sensing interface has been successfully fabricated.
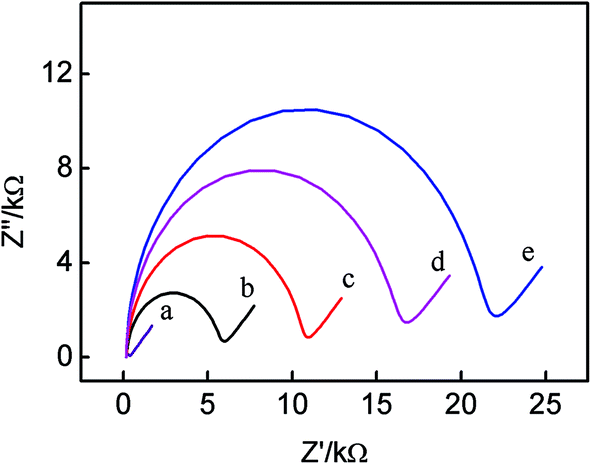 | ||
| Fig. 1 Electrochemical impedance spectra of the different modified electrodes in 0.1 M KCl solution containing 5 mM (1:1) [Fe(CN)6]3−/4−. (a) Bare Au electrode, (b) Au/DNA1 electrode, (c) Au/DNA1/MCH electrode, (d) Au/DNA1/MCH/DNA2 electrode, (e) Au/DNA1/MCH/DNA2/DNA3-MB electrode. The impedance spectra are obtained at the frequency range from 100 mHz to 100 kHz with a DC potential of +0.224 V. The amplitude is 5 mV. | ||
3.3. Feasibility of the biosensor
This present biosensing interface is expected to be used for the parallel analysis and continuous detection of two targets just as shown in Scheme 1, respectively. In order to prove the feasibility of this biosensor, the electrochemical responses of the different modified electrode were investigated, as illustrated in Fig. 2. Comparing with the Au/DNA1/MCH/DNA2 electrode (curve a), the Au/DNA1/MCH/DNA2/DNA3-MB electrode has an oxidation peak of MB at ca. −0.27 V (curve b),31,33 which further suggested the successful construction of the Au/DNA1/MCH/DNA2/DNA3-MB electrode. After the Au/DNA1/MCH/DNA2/DNA3-MB electrode was immersed into 100 nM MUC1 solution, the electrochemical oxidation current of MB greatly increased from 0.04 μA to 0.13 μA (curve c). This can be explained that after MUC1/aptamer complex was dissociated from the electrode, the remaining DNA1/DNA3-MB may be relatively flexible, and then MB was confined near the electrode, giving an enhanced electrochemical signal. On the other hand, when the Au/DNA1/MCH/DNA2/DNA3-MB was immersed into the CEA solution (100 ng mL−1), it can be observed that the electrochemical signal of MB decreased (curve d), and the corresponding electrochemical response (ΔIMB) was about 0.01 μA. It can be due to that the DNA3-MB/CEA complex was dissociated from the electrode and moved into the solution.3 Therefore, it can be noted that MB acted as a signal-on probe for MUC1, whereas a signal-off probe in biosensor for CEA. It demonstrates that the present biosensing interface is effective and useful for parallel analysis of two protein biomarkers.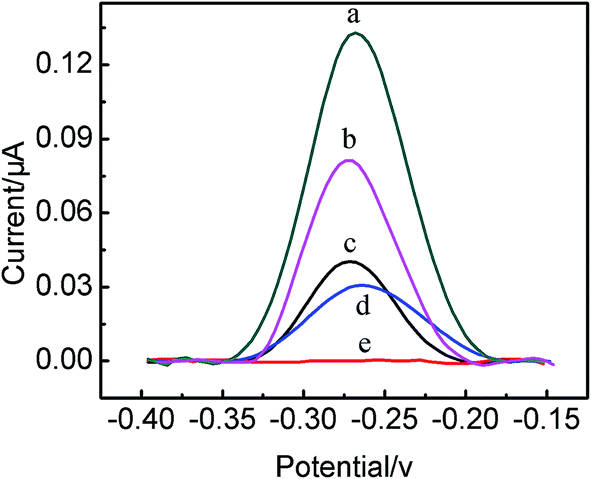 | ||
| Fig. 2 SWV curves of the different electrodes. (a) Au/DNA1/MCH/DNA2 electrode, (b) Au/DNA1/MCH/DNA2/DNA3-MB electrode, (c) Au/DNA1/MCH/DNA2/DNA3-MB electrode after incubated with 100 nM MUC1, (d) Au/DNA1/MCH/DNA2/DNA3-MB after treated incubated 100 ng mL−1 CEA, (e) electrode of (c) further incubated with 100 ng mL−1 CEA. | ||
More significantly, after the Au/DNA1/MCH/DNA2/DNA3-MB was employed to detect MUC1, and the obtained electrode was continuously used for the detection of CEA (100 ng mL−1) (curve e), and it was found that the electrochemical response decreased from ca. 0.13 μA to ca. 0.08 μA (curve e). The corresponding electrochemical response (ΔIMB) was 0.05 μA, which was five times higher than that for the parallel detection of CEA, resulting in the amplification of the CEA detection sensitivity. Therefore, it indicated that the continuous detection of two protein biomarker using the same sensing interface can be carried out, and even the detection of one target (taking MUC1 as a model) can enhance the detection sensitivity of another target (e.g. CEA). This present strategy for the continuous detection of targets may pave a new way for the development of the multifunctional biosensor.
3.4. Optimization of analytical conditions
It has been reported that the surface probe density has a key effect on the performance of aptasensor.34 In order to make sure the excellent performance of the biosensor, the amount of immobilized DNA on the gold electrode should be optimized. For this biosensor, DNA1 was firstly modified onto the bare gold electrode, the more assembled DNA1, the better for the subsequent modification of surface probe.26 Fig. 3A shows that the influence of different concentrations of DNA1 on the assembled amount of DNA1 onto the gold electrode. It can be observed that with the increase in DNA1 concentration, the electrochemical redox peaks of the DNA1 modified Au electrode in Fe[(CN)6]3−/4− solution decrease up to 10 μM. The higher DNA1 concentration almost cannot lead to the change of electrochemical redox peaks. Therefore, 10 μM DNA1 was chosen to modify the bare gold electrode. | ||
| Fig. 3 (A) Cyclic voltammograms of the different Au electrodes modified by DNA1 with different concentrations (from (a) to (e): 0 μM, 5 μM, 7.5 μM, 10 μM, 20 μM) in 0.1 M KCl solution containing 5 mM (1:1) [Fe(CN)6]3−/4−. (B) The current responses of the Au/DNA1/MCH/DNA2/DNA3-MB electrode in PBS with different pH values. | ||
Both the stability of DNA and the electrochemical response of MB are pH-dependent, therefore, the influence of the pH value on the electrochemical response of the Au/DNA1/MCH/DNA2/DNA3-MB electrode was investigated, as shown in Fig. 3B. From Fig. 3B, the maximum current response of the Au/DNA1/MCH/DNA2/DNA3-MB electrode can be observed at pH ∼ 7.0. Therefore, the solution of pH = 7.0 was selected as the optimal supporting electrolyte.
3.5. The electrochemical detection of two biomarkers
The parallel detection of MUC1 and CEA was carried out, and the corresponding results were shown in Fig. 4. Fig. 4A shows the electrochemical responses of the Au/DNA1/MCH/DNA2/DNA3-MB electrode after being treated with different concentration of MUC1. It can be seen that the oxidation peak current of MB at the modified electrode increased with MUC1 concentration increasing. It results from the reaction between DNA2 and MUC1 taking place, and the MUC1/aptamer complex released from the electrode, leaving the single-stranded DNA1/DNA3-MB folded. And thus, the electron transfer of MB was facilitated due to the shortened distance between the redox moiety and the electrode surface. The resulting calibration plots were shown in Fig. 4B and exhibited linear ranges from 10 nM to 100 nM for MUC1. And the corresponding linear function was ΔIMB = 0.00886cMUC1 (nM) + 0.0356 (r = 0.9989) with the detection limit of 0.6 nM (S/N = 3). The linear response range relatively wider than or comparable to that reported in previous aptasensor for MUC1.35,36 On the other hand, the sensing interface Au/DNA1/MCH/DNA2/DNA3-MB was used to detect CEA, and the corresponding results were shown in Fig. 4C and D. It displayed that the current response decreased linearly with the increase of the CEA concentration from 30 ng mL−1 to 300 ng mL−1, and the current response of the modified electrode decreased as a function of CEA concentrations. It can be obtained that the linear curve for CEA sensing was ΔIMB = 0.00069cCEA (ng mL−1) + 0.06204 (r = 0.9959) with the detection limit of 2.75 ng mL−1.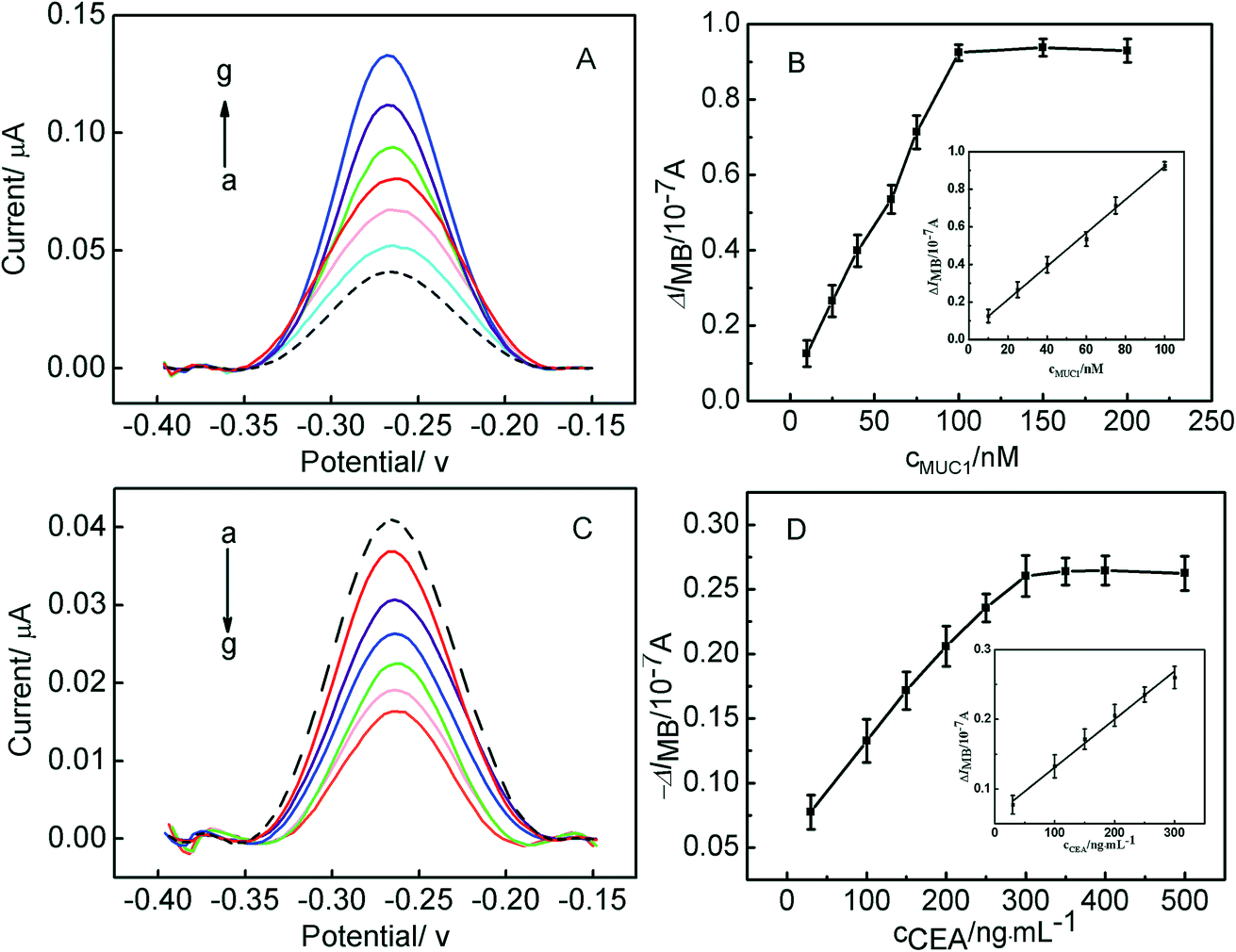 | ||
| Fig. 4 (A) SWV curves of the Au/DNA1/MCH/DNA2/DNA3-MB electrode for the detection of different concentrations of MUC1. The concentrations (from (a) to (g)) are 0 nM, 10 nM, 25 nM, 40 nM, 60 nM, 75 nM, 100 nM. (B) Calibration curve of the response current of the Au/DNA1/MCH/DNA2/DNA3-MB electrode to MUC1 in PBS (inset: the linear plot of the MB peak currents as functions of the concentrations of MUC1 from 10 nM to 100 nM). (C) SWV curves of the Au/DNA1/MCH/DNA2/DNA3-MB electrode for the detection of different concentrations of CEA. The concentrations (from (a) to (g)) are 0 ng mL−1, 30 ng mL−1, 100 ng mL−1, 150 ng mL−1, 200 ng mL−1, 250 ng mL−1, 300 ng mL−1. (D) Calibration curve of the response current of the Au/DNA1/MCH/DNA2/DNA3-MB electrode to CEA in PBS (inset: the linear plot of the MB peak currents as functions of the concentrations of CEA). | ||
More significantly, the sensing interface after being treated with MUC1 can be further employed for the detection of CEA. Herein, the Au/DNA1/MCH/DNA2/DNA3-MB electrode was treated with 100 nM MUC1, afterwards, the obtained electrode was used to measured CEA, the corresponding results has been shown in Fig. 5A. Fig. 5A displayed the relationship between the current response of the electrode and CEA concentration, it can be observed that the current responses decreased linearly with CEA concentration increasing from 10 ng mL−1 to 300 ng mL−1, and the corresponding linear function was ΔIMB = 0.00281cCEA (ng mL−1) + 0.23833 (r = 0.9965) with the detection limit of 0.5 ng mL−1 (S/N = 3). Furthermore, it can be obtained that the detection limits of this aptasensor for MUC1 and CEA is better than or comparable to those reported in literature.37,38 It further demonstrates that the continuous detection of two targets can be realized based on the present strategy, and the excellent detection sensitivity of CEA was obtained, which may be useful for the development of aptasensor in the clinical application.
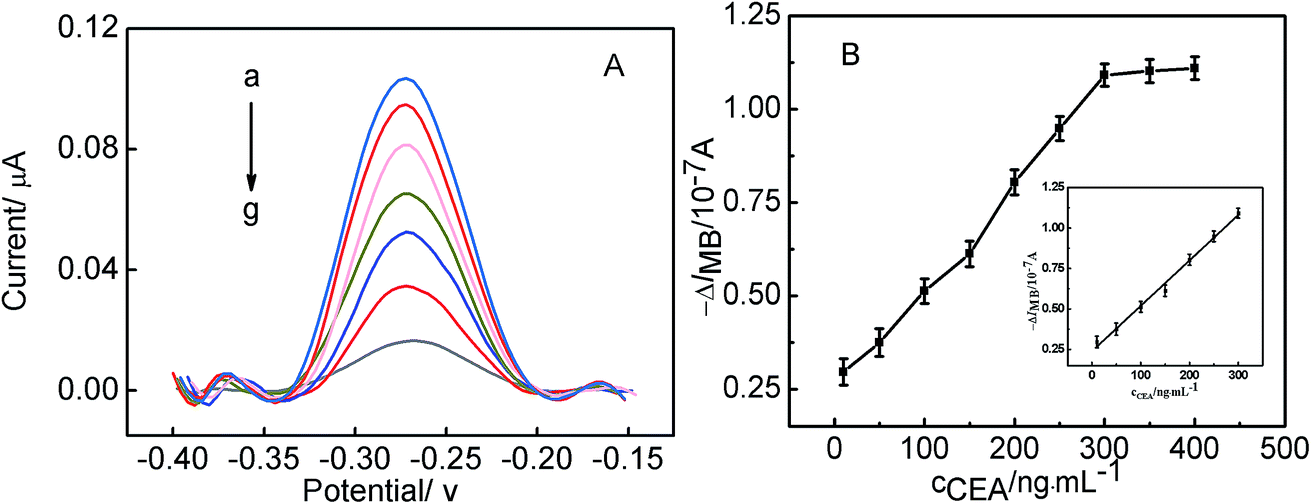 | ||
| Fig. 5 (A) SWV curves of the obtained electrode after the Au/DNA1/MCH/DNA2/DNA3-MB electrode treated with 100 nM MUC1 for the detection of different concentrations of CEA. The concentrations (from (a) to (g)) are 10 ng mL−1, 50 ng mL−1, (c) 100 ng mL−1, 150 ng mL−1, 200 ng mL−1, 250 ng mL−1, 300 ng mL−1. (B) Calibration curve of the response current of the obtained electrode after the Au/DNA1/MCH/DNA2/DNA3-MB electrode treated with 100 nM MUC1 to CEA in PBS (inset: the linear plot of the MB peak currents as functions of the concentrations of CEA). | ||
3.6. The regeneration, selectivity and stability of the aptasensor
For the parallel detection of MUC1 and CEA, the regeneration of this aptasensor was carried out by treated with 100 nM MUC1 or 300 ng mL−1 CEA, and then 20 μL DNA2 or DNA3-MB was dropped onto the electrode surface for 2 h. It can be seen from Fig. 6A and B that, after five cycles of the regeneration process, MB signals were almost recovered to the original values, which demonstrates that the fabricated biosensing interface can be regenerated in the parallel detection process. In case of the multiplexed detection of targets, the biosensing interface also can be regenerated by interacting with 100 nM MUC1 and 300 ng mL−1 CEA, as shown in Fig. 6C. It can be observed that, MB signals also recovered to the original values, which indicates that the designed aptasensor possesses excellent regeneration stability for the multiplexed detection of targets.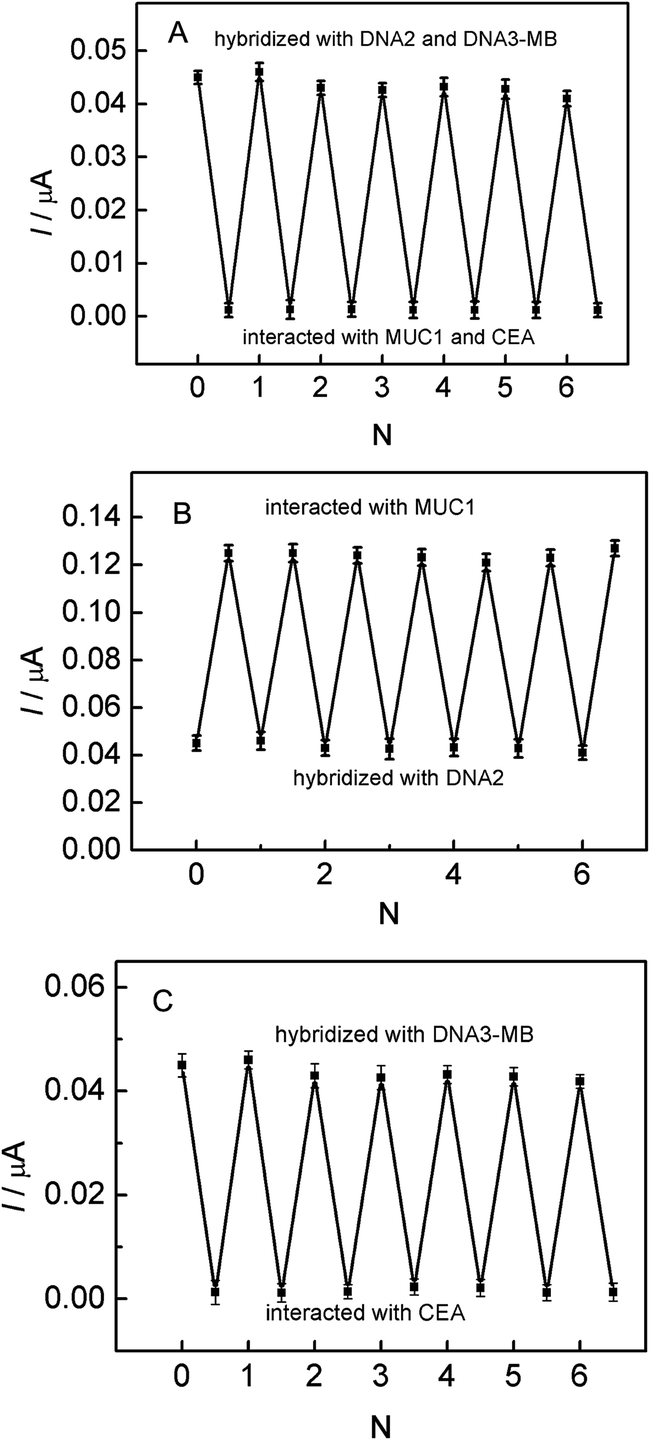 | ||
| Fig. 6 (A) The regeneration of this designed aptasensor by stepwise interaction with MUC1 (100 nM) and then hybridization of DNA2. (B) The regeneration of this designed aptasensor by stepwise interaction with CEA (300 ng mL−1) and then hybridization of the DNA3-MB. (C) The regeneration of this designed aptasensor by stepwise interaction with MUC1 (100 nM) and CEA (300 ng mL−1) and then hybridization of DNA2 and DNA3-MB. | ||
From the practical point, the specificity of the biosensor is an important feature. In order to evaluate the selectivity of the aptasensor, the interference of non-specific molecules in the detection of MUC1 or CEA was investigated and the results were illustrated in Fig. 7. It was found that the signal responses for 100 nM tumor necrosis factor-α (TNF-α), and 100 nM thrombin are much lower than those for MUC1 and CEA, which almost may be neglected.
 | ||
| Fig. 7 SWV curves of the Au/DNA1/MCH/DNA2/DNA3-MB in 10 mM PBS at ph 7.4 before (b) and after incubation with 100 nM MUC1 (a), 100 nM thrombin (c), 100 ng mL−1 CEA (e), 100 nM TNF-α (d). | ||
Additionally, the storage stability of the present aptasensor was also investigated, and it was found that the aptasensor remains bioactive after a two-week storage at 4 °C in a refrigerator, and the response currents have no significant change. These results demonstrate that the developed biosensor has acceptable stability. The reproducibility of the Au/DNA1/MUC1/DNA2/DNA3-MB electrode was also investigated. Five modified electrode were used to detect MUC1, and the relative standard deviation was 4.3%, indicating acceptable reproducibility of the aptasensor.
3.7. Recovery test
The recovery experiments for different MUC1 and CEA concentrations were carried out to evaluated applicability and reliability of the developed biosensor in a complex system. Blood serum samples were employed in this work. As shown in Table 1, the recoveries for the added MUC1 with 5, 25, 50, and 100 nM are 104%, 96%, 93%, and 102%, respectively. And from Table 2, the recoveries for the added CEA with 10, 50, 100, 300 ng mL−1 are 102%, 104%, 91%, 93%, respectively. The standard deviations of measurements were taken from four independent experiments. From Tables 1 and 2, it can be seen the recovery are acceptable. This implies that the developed biosensor has a good accuracy, which also indicates a promising feature for the analytical application in complex biological samples.| Sample | Added (nM) | Found (nM) | Recovery (%) |
|---|---|---|---|
| 1 | 5 | 5.2 ± 0.24 | 104 |
| 2 | 25 | 24 ± 3.3 | 96 |
| 3 | 50 | 46.5 ± 9.6 | 93 |
| 4 | 100 | 102 ± 12 | 102 |
| Sample | Added (ng mL−1) | Found (ng mL−1) | Recovery (%) |
|---|---|---|---|
| 1 | 10 | 10.2 ± 0.5 | 102 |
| 2 | 50 | 52 ± 4.6 | 104 |
| 3 | 100 | 91 ± 17 | 91 |
| 4 | 300 | 278 ± 32 | 93 |
4. Conclusions
In summary, based on the novel biosensing strategy, a simple and regenerable electrochemical aptasensor for the parallel and the continuous detection of two targets was fabricated. Herein, not only the parallel analysis of MUC1 and CEA can be carried out, but also the continuous detection of MUC1 and CEA using the same interface was realized. And moreover, the excellent analytical performance for the detection of MUC1 and CEA was obtained. So, according to the requirements in analytical application, the different protocols given in this work can be selected. More significantly, the sensing interface can be simply regenerated after the parallel or continuous detection of targets. We believe that this designed concept could promote the development of aptasensor in the field of the multiplexed detection of biomarkers and also the application of the biosensor will be expanded. This present work may have very high and potential values for basic researches as well as clinical applications in the future.Acknowledgements
This work was financially supported by the National Nature Science Foundation of China (21005090, 21273288, 21573290), and Hunan Provincial Natural Science Foundation of China (13JJ3004) and the China Postdoctoral Science Foundation (2012M510136, 2013T60774), the National High Technology Research and Development Program of China (2015AA020502).References
- A. J. Atkinson, W. A. Colburn, V. G. DeGruttola, D. L. DeMets, G. J. Downing, D. F. Hoth, J. A. Oates, C. C. Peck, R. T. Schooley and B. A. Spilker, Biomarkers and Surrogate Endpoints: Preferred Definitions and Conceptual Framework, Clin. Pharmacol. Ther., 2001, 69, 89–95 CrossRef PubMed.
- R. Hu, W. Wen, Q. L. Wng, H. Y. Xiong, X. H. Zhang, H. S. Gu and S. F. Wang, Biosens. Bioelectron., 2014, 53, 384–389 CrossRef CAS PubMed.
- T. Xu, N. Liu, J. Yuan and Z. F. Ma, Biosens. Bioelectron., 2015, 70, 161–166 CrossRef CAS PubMed.
- B. V. Chikkaveeraiah, A. A. Bhird, N. Y. Morgan, H. S. Eden and X. Y. Chen, ACS Nano, 2012, 6, 6546–6561 CrossRef CAS PubMed.
- M. Hu, J. Yan, Y. He, H. T. Lu, L. X. Weng, S. P. Song, C. H. Fan and L. H. Wang, ACS Nano, 2010, 4, 488–494 CrossRef CAS PubMed.
- Z. X. Jiang, Y. Qin, Z. Peng, S. H. Chen, S. Chen and C. Y. Deng, Biosens. Bioelectron., 2014, 62, 268–273 CrossRef CAS PubMed.
- F. Qu, H. M. Lu, M. H. Yang and C. Y. Deng, Biosens. Bioelectron., 2011, 26(12), 4810–4814 CrossRef CAS PubMed.
- S. Boddington, T. D. Henning, E. J. Sutton and H. E. Daldrup-Link, J. Visualized Exp., 2008, 14, 686 Search PubMed.
- S. Choi, J. H. Yu, S. A. Patel, Y. L. Tzeng and R. M. Dickson, Photochem. Photobiol. Sci., 2011, 10, 109–115 Search PubMed.
- T. K. Tsukagoshi, T. Nakamura and R. Nakajima, Anal. Chem., 2002, 74(16), 4109–4116 CrossRef PubMed.
- D. J. Buchsbaum, Cancer, 1997, 80(12), 2371–2377 CrossRef CAS PubMed.
- J. Liu, H. Zhou, J. J. Xu and H. Chen, Chem. Commun., 2011, 47, 4388–4390 RSC.
- J. J. Yin, X. X. He, K. M. Wang, F. Z. Xu, J. F. Shangguan, D. G. He and H. Shi, Anal. Chem., 2013, 85, 12011–12019 CrossRef CAS PubMed.
- D. L. Robertson and G. F. Joyce, Natrue, 1990, 344(6265), 467–468 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346(6287), 818–822 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249(4968), 505–510 CrossRef CAS PubMed.
- Y. He, Y. Q. Chai, H. J. Wang, L. J. Bai and R. Yuan, RSC Adv., 2014, 4, 56756–56761 RSC.
- Y. F. Huang, H. T. Chang and W. Tan, Anal. Chem., 2008, 80, 567–572 CrossRef CAS PubMed.
- X. Liu, Y. Qin, C. Y. Deng, J. Xiang and Y. J. Li, Talanta, 2015, 13, 150–154 CrossRef PubMed.
- S. Q. Zhang, K. Wang, J. L. Li, Z. Y. Li and T. Sun, RSC Adv., 2015, 5, 75746–75752 RSC.
- D. N. Jiang, F. Liu, L. Q. Zhang, L. L. Liu, C. Liu and X. Y. Pu, RSC Adv., 2014, 4, 57064–57070 RSC.
- J. J. Li, X. Q. Zhong, H. Q. Zhang, X. C. Le and J. J. Zhu, Anal. Chem., 2012, 84(12), 5170–5174 CrossRef CAS PubMed.
- Y. Du, C. G. Chen, M. Zhou, S. J. Dong and E. K. Wang, Anal. Chem., 2011, 83, 1523–1529 CrossRef CAS PubMed.
- S. Manna, S. Senapati, S. Lindsay, P. M. Zhang and J. Am, Chem. Soc., 2015, 137(23), 7415–7423 CrossRef CAS PubMed.
- S. J. Lee, B. Alder, S. Ekstrom, M. Rezeli, A. Vegvari, J. W. Park, J. Malm and T. Laurell, Anal. Chem., 2014, 86(15), 7627–7634 CrossRef CAS PubMed.
- H. Y. Wang, W. Gong, Z. Tan, X. X. Yin and L. Wang, Electrochim. Acta, 2012, 76, 416–423 CrossRef CAS.
- Y. Du, B. L. Li, H. Wei, Y. L. Wang and E. K. Wang, Anal. Chem., 2008, 80, 5110–5117 CrossRef CAS PubMed.
- C. Y. Deng, J. H. Chen, L. H. Nie and S. Z. Yao, Anal. Chem., 2009, 81(24), 9972–9978 CrossRef CAS PubMed.
- Y. Li, L. Deng, C. Y. Deng, Z. Nie, M. H. Yang and S. Si, Talanta, 2012, 99, 637–642 CrossRef CAS PubMed.
- Y. Cao, S. Zhu, J. C. Yu, X. J. Zhu, Y. M. Yin and G. X. Li, Anal. Chem., 2012, 84, 4314–4320 CrossRef CAS PubMed.
- L. Wu, X. H. Zhang, W. Liu, E. H. Xiong and J. H. Chen, Anal. Chem., 2013, 85(17), 8397–8402 CrossRef CAS PubMed.
- D. Xu, D. Xu, X. Yu, Z. Liu, W. He and Z. Ma, Anal. Chem., 2005, 77(16), 5107–5113 CrossRef CAS PubMed.
- F. F. Yan, F. Wang and Z. L. Chen, Sens. Actuators, B, 2011, 160(1), 1380–1385 CrossRef CAS.
- S. Zhao, W. Yang and R. Y. Lai, Biosens. Bioelectron., 2011, 26(5), 2442–2447 CrossRef CAS PubMed.
- X. Liu, Y. Qin, C. Y. Deng, J. Xiang and Y. J. Li, Talanta, 2015, 132, 150–154 CrossRef CAS PubMed.
- F. Ma, C. Ho, A. K. H. Cheng and H. Z. Yu, Electrochim. Acta, 2013, 110, 139–145 CrossRef CAS.
- R. Hu, W. Wen, Q. L. Wang, H. Y. Xiong, X. H. Zhang, H. S. Gu and S. F. Wang, Biosens. Bioelectron., 2014, 53, 384–389 CrossRef CAS PubMed.
- C. Y. Qin, Y. Gao, W. Wen, X. H. Zhang and S. F. Wang, Biosens. Bioelectron., 2016, 79, 522–530 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |