DOI:
10.1039/C6RA09148B
(Paper)
RSC Adv., 2016,
6, 63271-63285
Synthesis, structure, electrochemical, DNA interaction and antimicrobial studies of fluorinated trans-dicationic pyridinium porphyrins†
Received
9th April 2016
, Accepted 22nd June 2016
First published on 23rd June 2016
Abstract
A series of trans-pyridyl porphyrins, 5,15-di(pentafluorophenyl)-10,20-bis(2′/3′/4′-pyridyl)porphyrin (1–3), trans-dicationic pyridinium porphyrins, 5,15-di(pentafluorophenyl)-10,20-bis(2′/3′/4′-N-methylpyridyl)porphyrins (4–6) and their copper(II) and zinc(II) derivatives were synthesized. These compounds were characterized using various spectroscopic methods, electrochemical and single crystal X-ray crystallographic studies. The trans-dicationic porphyrin derivatives exhibit red shifted absorption spectra over the simple pyridyl porphyrins. The reduction potentials of trans-pyridyl porphyrins are more positive than those of MTPPs. Crystal structure of 3c is forming networks of molecules through Zn–N coordination displaying large number of channels. The intermolecular interactions involving fluorine contributes considerably to the crystal packing in all the structures which was confirmed by computational Hirshfeld surface analysis. The dicationic porphyrins were further explored for its DNA interaction abilities and antimicrobial activities. The UV-visible and fluorescence spectroscopic titrations indicate that the porphyrins bind with the calf thymus DNA by outside groove binding mode with or without self-stacking. The intrinsic binding constants Kb of these dicationic porphyrins to DNA was found to be in the range of 105 to 106 M−1. The results reveal that among the three sets of porphyrins (4–6), the 3-/4-pyridyl derivatives display higher DNA binding activities compared to the 2-pyridyl analogues. The photocleavage experiments disclose that the porphyrins employ 1O2-mediated mechanism in cleaving DNA and the freebase and zinc(II) derivatives show better photoinduced cleavage ability compared to its copper(II) analogues. The dicationic porphyrins also show significant antimicrobial activities than those of non-fluorinated analogues.
Introduction
Synthetic molecules that interact efficiently with nucleic acids are well known as artificial nucleases. They are highly stable, low cost and site specific alternatives to natural nucleases which help to improve the essential understanding of mechanistic characteristics of enzyme action, to develop new biotechnological tools and also in nucleic acid targeting therapeutics. Porphyrin based artificial nucleases are very important due to their application in photodynamic therapy (PDT).1,2 They are fascinating molecules because of their unique spectroscopic, geometric and physicochemical properties and also find use in various fields including material chemistry and biomimetics.3 In particular, cationic porphyrins enter the cells readily and mainly interact with DNA through three major binding modes involving intercalation, outside groove binding and outside binding without self-stacking depending on the DNA base composition, choice of peripheral substituents and the nature of central metal on the porphyrin.4 When porphyrins are photo activated, singlet oxygen (1O2) is produced by energy transfer from the photo excited sensitizer to the ground state oxygen which is known as type II mechanism and it is more important in PDT. The excited photosensitizer can act as a reducing agent in the formation of reactive oxygen species such as superoxide radical, hydrogen peroxide or hydroxyl radical.3
Fiel and coworkers first investigated the interaction of cationic porphyrins with the negatively charged backbone of DNA either by electrostatic binding or intercalation.5 The cationic porphyrins act as a bifunctional compounds and due to the electrostatic interaction between the positively charged porphyrin periphery and anionic phosphate backbone of nucleic acid, effective interaction with DNA is possible. And, the binding modes can be studied using UV-visible titration, fluorescence, viscometry etc. and the DNA cleavage ability is usually detected by agarose gel electrophoresis.6–9 Porphyrins bearing pyridyl/pentafluorophenyl substituents at the porphyrin periphery have been widely used as photosensitizer in PDT due to their high triplet quantum yields and also efficient catalyst for various redox reactions.10–12 The pyridyl substituted porphyrins can also act as important building blocks for supramolecular self-assemblies.13,14 Also, fluorinated porphyrins show enhanced pharmacokinetic properties compared to its non-fluorinated analogues and in fact they mimic the biological functions of enzyme cytochrome P450.15–17 In order to explore the combined properties of a porphyrin containing both pentafluorophenyl group as well as pyridyl group at the porphyrin periphery, we synthesized the porphyrins namely, trans-pyridyl porphyrins, 5,15-di(pentafluorophenyl)-10,20-bis(2′/3′/4′-pyridyl)porphyrin (1–3), trans-dicationic pyridinium porphyrins, 5,15-di(pentafluorophenyl)-10,20-bis(2′/3′/4′-N-methylpyridyl)porphyrins (4–6) and their copper(II) and zinc(II) derivatives using 5-(pentafluorophenyl)dipyrromethane and 2-/3-/4-pyridine carboxaldehyde as a precursors (Fig. 1). Their photophysical, electrochemical properties, DNA interaction abilities as well as its antimicrobial activities have also been explored.
 |
| | Fig. 1 Chemical structures of trans-pyridyl and trans-dicationic pyridinium porphyrins under study. | |
Experimental
Materials and methods
Analytical grade reagents/chemicals were purchased and used as such without any further purification. Solvents were purified as per the standard procedures18 before its use for any chemical reaction. Calf thymus DNA was purchased from Sigma Aldrich. The supercoiled pBR 322 DNA and ethidium bromide was purchased from Merck. UV-visible spectra were recorded at room temperature in DMF using a Shimadzu UV-2450 double beam spectrophotometer and 1H NMR spectra were taken in a Bruker Avance III 400 MHz spectrometer. Mass spectra were recorded under ESI/HR-MS at 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 resolution using Thermo Scientific Exactive mass spectrometer (Thermo Fischer Scientific, Bremen, Germany). Fluorescence spectra were measured using a Perkin Elmer LS 55 luminescence spectrophotometer. Fluorescence lifetime measurements were calculated from time-resolved fluorescence intensity decays using Horiba Scientific Fluoromax-4 spectrofluorometer with data station software in time correlated single photon counting mode. Cyclic voltammetric measurements of the porphyrin samples were performed on a CH-instruments Inc, USA (Model: CHI660E) equipped with potentiostat/galvanostat with Fourier Transform AC Voltammeter. The electrochemical cell consists of a three-electrode cell assembly; a glassy carbon working electrode, standard calomel electrode (SCE) as reference electrode and a platinum wire as the auxiliary electrode. The concentrations of the porphyrin samples were employed as ∼0.5 mM. All the measurements were performed at 25 °C in DMF under N2 atmosphere using 0.1 M tetrabutylammonium hexafluorophosphate (TBAHFP) as the supporting electrolyte. Single crystal structure X-ray data collections were performed on a Bruker AXS Kappa Apex II CCD diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at 298 K.
800 resolution using Thermo Scientific Exactive mass spectrometer (Thermo Fischer Scientific, Bremen, Germany). Fluorescence spectra were measured using a Perkin Elmer LS 55 luminescence spectrophotometer. Fluorescence lifetime measurements were calculated from time-resolved fluorescence intensity decays using Horiba Scientific Fluoromax-4 spectrofluorometer with data station software in time correlated single photon counting mode. Cyclic voltammetric measurements of the porphyrin samples were performed on a CH-instruments Inc, USA (Model: CHI660E) equipped with potentiostat/galvanostat with Fourier Transform AC Voltammeter. The electrochemical cell consists of a three-electrode cell assembly; a glassy carbon working electrode, standard calomel electrode (SCE) as reference electrode and a platinum wire as the auxiliary electrode. The concentrations of the porphyrin samples were employed as ∼0.5 mM. All the measurements were performed at 25 °C in DMF under N2 atmosphere using 0.1 M tetrabutylammonium hexafluorophosphate (TBAHFP) as the supporting electrolyte. Single crystal structure X-ray data collections were performed on a Bruker AXS Kappa Apex II CCD diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at 298 K.
Synthesis of trans-porphyrins
Synthesis of 5,15-di(4′-pyridyl)-10,20-bis(pentafluorophenyl)porphyrin and their metal derivatives. The precursor, 5-(pentafluorophenyl)dipyrromethane was prepared using the available literature method.19 5-(Pentafluorophenyl)dipyrromethane (0.1 g, 3.2 × 10−4 mol) and 4-pyridinecarboxaldehyde (0.034 g, 43.2 × 10−4 mol) were added to a dry double neck round-bottom flask with 350 mL of CH2Cl2 and N2 gas purged for 10 min. Then, trifluoroacetic acid (TFA, 1.5 mL) was added and the contents were stirred under N2 at ice cold temperature (∼5 °C) for 30 min. To that, about 0.14 g of 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) was added and the reaction mixture was stirred at room temperature for an additional 1 h. After that, the solution was quenched with saturated NaHCO3 and extracted with CHCl3. Compound was purified by silica column chromatography using chloroform–acetone mixture and was dried under vacuum. The porphyrin, 3a was yielded as a purple solid (0.046 g; yield: 36%). The copper(II) (3b) and zinc(II) (3c) complexes were synthesized in chloroform/methanol mixture using the corresponding metal acetate and the yields were found to be quantitative (93–95%). UV-visible data of porphyrins in DMF at 298 K, λmax (logε/M−1 cm−1): 3a, 412 (5.41), 507 (4.21), 582 (3.71); 3b, 411 (5.40), 537 (4.13); 3c, 421 (5.24), 553 (3.85). 1H NMR data of 3a (400 MHz, CDCl3): δ (ppm) 9.08 (d, 4H), 8.91 (d, 4H), 8.87 (d, 4H), 8.17 (d, 4H), −2.92 (s, 2H). ESI mass data of 3a (C42H18N46F10): 796.62 (calcd), 797.15 (found). Fluorescence spectral data of porphyrins in DMF at 298 K (λex at 415/416 nm), λem for 3a: 640 (s), 706 (s); λem for 3c: 600 (s), 649 (s). Similarly, the 2′-/3′-pyridyl and its metal complexes (1a–1c, 2a–2c) were synthesized and characterized. 1H NMR data of 1a (300 MHz, CDCl3): δ (ppm) 9.17 (d, 2H), 8.97 (d, 4H), 8.85 (d, 4H), 8.28 (d, 2H), 8.17 (q, 2H), 7.77 (m, 2H), −2.84 (s, 2H). 1H NMR data of 2a (400 MHz, DMSO-d6): δ (ppm) 9.49 (d, 2H), 9.37 (d, 4H), 9.14 (q, 2H), 9.02 (d, 4H), 8.78 (d, 2H), 7.97 (m, 2H), −2.99 (s, 2H).
Synthesis of 5,15-di(4′-N-methylpyridyl)-10,20-bis(pentafluorophenyl)porphyrin and their metal derivatives. 5,15-Di(pyridyl)-10,20-bis(pentafluorophenyl)porphyrin (0.020 g, 2.5 × 10−5 mol) was taken in a dry double neck round-bottom flask containing 30 mL of CHCl3. Methyl trifluoromethanesulfonate (23 μL, 2 × 10−4 M) in 5 mL of CHCl3 was added in drops for 5 min and the contents were stirred vigorously for another 30 min. The precipitate was filtered through sintered crucible (G4) and thoroughly washed with CHCl3 to remove any other impurities. The desired product, 6a was obtained in 92% yield (0.026 g). The copper(II) (6b) and zinc(II) (6c) complexes were synthesized as discussed earlier and the yields were 93–95%. UV-visible data of porphyrins in DMF at 298 K, λmax (logε/M−1 cm−1): 6a, 417 (5.36), 509 (4.29), 583 (3.85); 6b, 416 (5.38), 541 (4.26); 6c, 426 (5.21), 557 (4.13). 1H NMR data (400 MHz, acetone-d6): δ (ppm), 6a, 9.64–9.09 (m, 16H), 5.00 (s, 6H), −2.95 (s, 2H); 6c, 9.57 (s, 4H), 9.93 (s, 4H), 9.10 (d, 8H), 4.98 (s, 6H). Fluorescence spectral data of porphyrins in DMF at 298 K (λex at 415/416 nm), λem for 6a: 646 (s), 707 (s); λem for 6c: 616 (s), 654 (s). ESI-mass data of 6a: 811.17, loss of two triflate anions and a methyl group (calcd: 1124.82). Similarly, the porphyrins, 4a–4c and 5a–5c were synthesized and characterized. 1H NMR data of 4a (400 MHz, acetone-d6): 9.63 (d, 2H), 9.47 (s, 4H), 9.19 (s, 4H), 8.97 (m, 4H), 8.72 (t, 2H), 4.06 (d, 6H), −3.07 (s, 2H). 1H NMR data of 5a (400 MHz, DMSO-d6): δ (ppm) 10.06 (d, 2H), 9.55 (d, 2H), 9.46 (s, 6H), 9.25 (s, 4H), 8.62 (t, 2H), 4.87 (s, 6H), −3.16 (s, 2H).
Single crystal X-ray structure determination
Upon mounting the appropriate sizes of the single crystals of porphyrins (1a, 2a, 3c, 6a and 6c) on a glass capillary, the crystal structure data collections were achieved on a Bruker AXS Kappa Apex II CCD diffractometer. The reflections with I > 2σ(I) were employed for structure solution and refinement. The structure was solved using SIR92 (ref. 20) (WINGX32) program by direct methods. And, structure refinement was done using the SHELXL97 (ref. 21) software by successive Fourier synthesis on |F|2 using the full-matrix least squares refinement.
Hirshfeld surface analysis
To analyse the intermolecular interactions present in the crystals, Hirshfeld surface analysis and generation of 2D fingerprint plots were obtained using Crystal Explorer 3.1 (ref. 22) based on results of single crystal X-ray diffraction analysis.
DNA binding studies
The concentration of the calf thymus (CT) DNA was determined using the average extinction coefficient value of 6600 M−1 cm−1 of a single nucleotide at 260 nm.23 The stock solutions (100 mM) of compounds 4–6 were prepared in 6% DMF, 50 mM Tris buffer and 50 mM NaCl, diluted in millipore water for further use. Spectroscopic titrations were carried out to determine the binding affinity between DNA and porphyrins under study (4–6). Absorption titrations were carried out in the presence of fixed concentration of porphyrins (3 μM) and titrated with varying concentration of DNA (0–3 μM).24 The fluorescence emission spectra of freebase and zinc(II) dicationic porphyrins were recorded in the range of 450–800 nm (λexc = 411–426 nm). The change in fluorescence intensity was observed by titrating the fixed amount of cationic porphyrins (1 μM) with varying concentration of CT–DNA (0–1 μM).25 The relative binding of porphyrins to DNA (12 μM) was studied by fluorescence spectroscopy using ethidium bromide (EB) bound to DNA solution in Tris–buffer (pH = 7.0).26 The fluorescence intensity at 490–800 nm (λex = 480 nm) of EB (7.5 μM) with an increasing amount of the complex concentration (0–12 μM) was recorded. The final volume of the reaction mixture was made to 3 mL by adding 10 mM of Tris–HCl buffer (pH 7.2).
DNA cleavage experiments
The cleavage of supercoiled pBR 322 DNA was studied by agarose gel electrophoresis.27 The pBR 322 DNA (0.2 μg) was treated with the porphyrins of two different concentrations (5 and 10 μM) and the reactions were carried out in 50 mM Tris–HCl buffer (pH = 7.2, [NaCl] = 50 mM), and then the solutions were irradiated with a UV illuminator (365 nm, 15 W) at 25 °C for 60 min. The samples were then analyzed by electrophotolysis for 1–2 hours at 75 V on 1% agarose gel in TBE buffer. The gel was stained with 0.5 μg mL−1 ethidium bromide (EB) and photographed under UV light using Gel Doc 100 system, Bio-Rad; Hercules, CA. All experiments were performed at least triplicate.
Measurement of singlet oxygen
A steady-state method was implemented to find the singlet oxygen quantum yield,28 using 1,3-diphenylisobenzofuran (DPBF) as the oxygen scavenger and meso-tetrakis(tetraphenyl)porphyrin (H2TPP) as the reference. DPBF (50 μM) solutions without or with porphyrin derivatives (0.5 μM) were prepared in DMSO under dark conditions. These solutions were irradiated at room temperature and under gentle magnetic stirring. The breakdown of DPBF was monitored by measuring the decrease in absorbance at 410 nm at pre-established irradiation intervals. From this plot, the rates of 1O2 production of porphyrins under study relative to those of H2TPP were determined. Light intensities were measured with a pyroelectric detector (RJP-735 from Laser Probe) which was connected to an energy ratio meter (RJ-7620 from Laser Probe) and the average light fluence rate was 95 μJ cm−2. The singlet oxygen quantum yield29,30 of the porphyrins with respect to the reference was calculated using the equation:
| ϕ(1O2)comp = ϕ(1O2)ref(mcomp/mref)(Fref/Fcomp) |
where m is the slope of a plot of difference in change in absorbance of DPBF (at 410 nm) with the irradiation time, and F is the absorption correction factor (F = 1 − 10−OD).
Antimicrobial and cytotoxicity assessment
The antimicrobial activity of the 4-pyridyl (3a–3c) and 4-N-methylpyridyl porphyrins (6a–6c) were performed using the literature method31 against the MTCC cultures which were obtained from Microbial culture collection, Chandigarh, India are as follows: two Gram-positive Staphylococcus aureus, Bacillus subtilis and two Gram-negative Pseudomonas aeruginosa and Escherichia coli bacterial strains and a fungal strain Candida albicans were used. The ligand and their metal complexes were dissolved in dimethylsulphoxide (DMSO) to a stock solution of 210–560 μM. The sample at three different concentrations (40–160 μM) was loaded onto the sterile discs on agar plates directly. Plates swabbed with the bacteria culture were incubated at 35–37 °C for 24 hours. At the end of the incubation period, inhibition zones formed on the medium were evaluated in mm and studies were performed in duplicate. Solvent control test was also performed in order to study the effect of dimethylsulphoxide on the growth of microorganism and it showed no activity against microbial strains. The standard antimicrobial drug (tetracycline) was also screened under similar condition for comparison.
Cytotoxicity of the tested compounds were performed by means of a colorimetric assay (MTT assay) using Vero (African green monkey epithelial kidney) cells, obtained from King Institute, Chennai, India. The assay was carried out after 48 h of cell incubation in the medium, containing compounds at concentrations ranging from 10 to 100 μg mL−1. The cells were cultured in open vented 25 cm3 culture flasks (Corning, NY) in a standard horizontal laminar flow chamber and incubated at 37 °C in an atmosphere of 5% CO2 and 95% air and were provided with the growth medium of 90% Dulbecco's Modified Eagle Medium (DMEM), 10% fetal bovine serum (Hi-Media®) and 5% streptomycin–penicillin (Sigma-Aldrich, USA). A quantity of 0.5–1 × 104 cells per mL was seeded into a 96-well plate and after 24 or 48 h, the cells were washed and maintained with different concentrations of samples (10–100 μg mL−1) and incubated for 48 h at 37 °C under 5% CO2 atmosphere. The extent of MTT reduction was measured spectrophotometrically at 570 nm and a reference at 630 nm with a microplate reader. The percentage of inhibition of the cells are measured using the formula,
The minimal inhibitory concentration (MIC) of compounds under antimicrobial study were determined using the standard method of Wariso and Ebong.32 The compounds were dissolved in DMSO, the final concentration of sample after dilution are in the range of 100–1.56 mg mL−1. The MIC value corresponds to the lowest concentration that inhibited the growth after 24 h at 37 °C.
Results and discussion
Consuming 5-(pentafluorophenyl)dipyrromethane and 2/3/4-pyridinecarboxaldehyde, the desired trans-pyridylporphyrins, 5,15-di(pentafluorophenyl)-10,20-bis(2′/3′/4′-pyridyl)porphyrin were synthesized using the modified procedure of Lindsey et al.33 and their copper(II) and zinc(II) metal complexes were prepared by the variant literature method.34
N-Methylation of trans-pyridyl porphyrins was performed using methyl trifluoromethanesulfonate in CHCl3 medium to yield the corresponding trans-dicationic pyridinium porphyrins. All the synthesized porphyrins (Scheme 1) were isolated, purified by column chromatography and characterized by spectral, electrochemical methods and single crystal X-ray diffraction analysis.
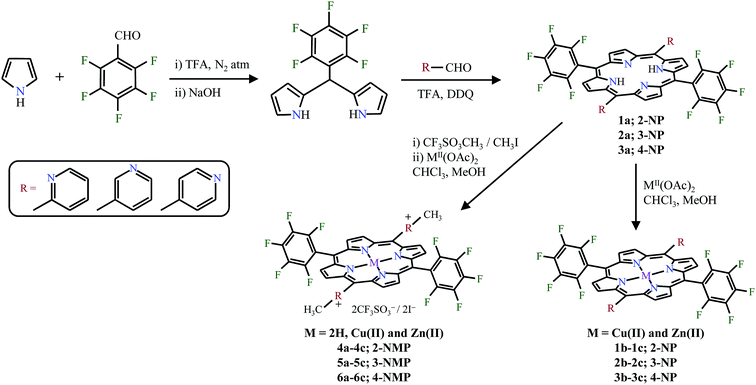 |
| | Scheme 1 Synthesis of trans-pyridyl porphyrins and its dicationic pyridinium derivatives. | |
Optical absorption data of porphyrins, 1–6 were measured in DMF, exhibited an intense Soret (B) and 2–4 visible (Q) bands and these absorption spectral features are similar to that of corresponding 5,10,15,20-tetraphenylporphyrins, TPPs.34 It is also observed that there is a marginal blue shifted absorption spectra for copper(II) complexes and considerable red shift of 8–10 nm for zinc(II) complexes compared to that of the free ligand (Fig. 2a).
 |
| | Fig. 2 (a) Optical absorption and (b) fluorescence emission spectra of 1a–6a in DMF at 298 K. | |
The fluorescence spectra of porphyrins, 1–6 were measured using DMF as solvent and Fig. 2b shows the fluorescence emission spectra of various trans-porphyrins under study. On excitation at the Soret band, the free ligands show two well defined emission peaks around 640–646 nm and 704–708 nm whereas the zinc(II) derivatives exhibit around 597–616 nm and 649–654 nm. Interestingly, the zinc(II) derivative of dicationic 4-pyridinium porphyrin, 6c shows a red shift of 16 nm compared to its free ligand 4-pyridylporphyrin, 3c (Table 1).
Table 1 Photophysical data of trans-pyridyl and trans-dicationic pyridinium porphyrins, 1–6 recorded in DMF
| Porphyrin |
Absorption, λmax (nm) |
Fluorescencea, λmax (nm) |
| 2H, a |
Cu(II), b |
Zn(II), c |
2H, a |
Cu(II)b, b |
Zn(II), c |
| Fluorescence spectra of the porphyrins were obtained as a function of λex in the Soret band. Less intense fluorescence peaks were observed for the Cu(II) complexes. |
| 1 |
412, 508, 583, 639 |
411, 537, 568 |
421, 552 |
642, 706 |
649, 710 |
597, 651 |
| 2 |
414, 509, 542, 584, 640 |
412, 539, 571 |
422, 553 |
642, 708 |
654, 712 |
602, 653 |
| 3 |
412, 507, 582 |
411, 537 |
421, 553 |
640, 706 |
649, 707 |
600, 649 |
| 4 |
415, 507, 583 |
414, 542, 574 |
425, 555 |
640, 704 |
650, 710 |
600, 651 |
| 5 |
416, 508, 583 |
415, 575 |
426, 556 |
639, 705 |
655, 711 |
597, 650 |
| 6 |
417, 509, 583 |
416, 541 |
426, 557 |
646, 707 |
653, 704 |
616, 654 |
Electrochemical studies
The redox potentials of trans-porphyrins, 1–6 were measured by cyclic voltammetry and compared to those of simple tetraphenylporphyrins, MTPPs [M = 2H, Cu(II) and Zn(II)] (Table 2). A representative cyclic voltammograms of selected porphyrins, 2a, 5a, 3c and 6c are shown in Fig. 3. The trans-pyridyl porphyrins, 1–3 undergo two reversible one electron reduction reaction, forming the anion and dianion similar to that of MTPP counter parts. As predicted, the reduction potentials of trans-pyridyl porphyrins are more positive when compared to MTPPs.32
Table 2 Comparison of reduction potentials (mV) for 1a–6c in DMF using 0.1 M TBAHFP at 25 °C
| Compound |
Redn (1) |
Redn (2) |
Redn (3) |
Redn (4) |
| Not soluble in DMF. First reduction was not observed. |
| H2TPP |
— |
— |
−1108 |
−1532 |
| CuTPP |
— |
— |
−1190 |
−1714 |
| ZnTPP |
— |
— |
−1335 |
−1726 |
| 1a |
— |
— |
−851 |
−1349 |
| 1b |
— |
— |
−990 |
−1492 |
| 1c |
— |
— |
−1183 |
−1594 |
| 2a |
— |
— |
−810 |
−1302 |
| 2b |
— |
— |
−943 |
−1453 |
| 2ca |
— |
— |
— |
— |
| 3a |
— |
— |
−799 |
−1263 |
| 3b |
— |
— |
−923 |
−1486 |
| 3c |
— |
— |
−1058 |
−1554 |
| 4a |
−543 |
−872 |
−1093 |
−1325 |
| 4b |
−687 |
−832 |
−986 |
−1579 |
| 4cb |
— |
−834 |
−987 |
−1252 |
| 5ab |
— |
−738 |
−1240 |
−1426 |
| 5bb |
— |
−800 |
−1215 |
−1663 |
| 5cb |
— |
−910 |
−1264 |
— |
| 6a |
−577 |
−703 |
−1284 |
−1457 |
| 6b |
−676 |
−780 |
−1319 |
−1556 |
| 6c |
−722 |
−879 |
−1469 |
−1680 |
 |
| | Fig. 3 Overlayed cyclic voltammograms of (a) 2a, 5a and (b) 3c, 6c in DMF containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAHFP) as the supporting electrolyte with a scan rate of 0.1 V s−1. | |
Among the pyridyl porphyrins, the 4-pyridyl derivatives show more positive value of 250–300 mV than 2-/3-pyridyl analogues (150–250 mV) reflecting the net electron richness of the porphyrin core. The trans-dicationic pyridinium porphyrins, 4–6 show four electron reduction which occurs at the conjugated π-ring system (−986 to −1680 mV) as well as at the methylpyridyl substituents (−577 to −910 mV).32 In the case of 4c and 5a–5c, only three electron reduction process is observed possibly attributed to solvent effect.33 Due to the presence of electron withdrawing/positively charged substituents at the porphyrin periphery, the possible oxidation process was not observed in the studied compounds.35,36
X-ray crystallographic studies
Single crystal XRD analysis of porphyrins, 1a, 2a, 3c, 6a and 6c was done using the suitable size of single crystals which were grown at room temperature using appropriate solvents and the data are given in Table 3. All the compounds were crystallized in monoclinic system with various space groups as listed in Table 3. The asymmetric unit of structures, 1a, 2a, 3c, 6a and 6c contain half molecule of porphyrin with no solvent molecule, five water molecules, one THF molecule, a CF3SO3− anion and a CF3SO3− anion respectively.
Table 3 X-ray crystal structure data of porphyrins, 1a, 2a, 3c, 6a and 6c
| |
1a |
2a |
3c |
6a |
6c |
| R1 = ∑||Fo| − |Fc||/∑|Fo|; Io > 2σ(Io). wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Empirical formula |
C42H18F10N6 |
C42H26F10N6O4 |
C50H32F10N6O2Zn |
C46H24F16N6O8S2 |
C48H30F16N6O8S2Zn |
| fw |
796.62 |
868.69 |
1004.18 |
1156.83 |
1252.27 |
| CCDC no. |
1434316 |
1443805 |
1021573 |
1058021 |
1041762 |
| Colour |
Black |
Brown |
Purple |
Purple |
Red |
| Crystal system |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
C2/c |
C2/c |
P21/n |
P21/n |
| a, Å |
15.113(3) |
31.4898(16) |
23.891(4) |
13.5879(17) |
9.1236(4) |
| b, Å |
10.4895(16) |
6.7009(3) |
13.986(4) |
11.0738(10) |
14.0611(7) |
| c, Å |
11.628(2) |
22.5181(12) |
14.697(3) |
17.130(2) |
19.6345(10) |
| α, (deg) |
90 |
90 |
90 |
90 |
90 |
| β, (deg) |
112.085(9) |
122.093(2) |
105.084(15) |
113.223(4) |
100.7510(10) |
| γ, (deg) |
90 |
90 |
90 |
90 |
90 |
| T (K) |
293(2) |
293(2) |
293(2) |
293(2) |
293(2) |
| λ, Å |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
0.71073 |
| Volume (Å3) |
1708.1(5) |
4025.4(4) |
4742(2) |
2368.7(5) |
2474.7(2) |
| Z |
4 |
4 |
4 |
2 |
2 |
| Dcalcd (mg m−3) |
1.549 |
1.433 |
1.407 |
1.622 |
1.681 |
| No. of unique reflections |
2397 |
3949 |
4170 |
4175 |
6110 |
| No. of parameters refined |
263 |
372 |
361 |
495 |
364 |
| GOF on F2 |
0.957 |
1.051 |
1.041 |
1.137 |
1.028 |
| R1a |
0.0595 |
0.0518 |
0.0545 |
0.0779 |
0.0547 |
| wR2b |
0.0718 |
0.1392 |
0.1472 |
0.1632 |
0.1371 |
Interestingly, the zinc(II) centre in 6c is hexa-coordinated with the inner core nitrogens and two methanolic oxygens in its apex positions. The lattice THF molecule in 3c shows the well-known envelope conformation as observed earlier.37 The ORTEP and molecular crystal packing diagrams of porphyrins are shown in Fig. 4 and 5 respectively. Remarkably, there are no interactions on pyridyl nitrogen with any other atom on neither of the phenyl ring nor in the porphyrin periphery of the structures 1a and 2a. Unlike the reported36 crystal structure of 5,15-di(4′-pyridyl)-10,20-bis(pentafluorophenyl)porphyrin having weak π–π interactions between the porphyrin macrocycles leading to form a π-stacking of molecules, 1a (2-pyridyl) and 2a (3-pyridyl) do not have such interactions, instead the porphyrin molecules in both structures are arranged nearly in an orthogonal fashion (Fig. 5a and b).
 |
| | Fig. 4 ORTEP diagrams of (a) 1a, (b) 2a, (c) 3c, (d) 6a and (e) 6c (thermal ellipsoids are shown at 30% probability level; solvent molecules were not shown for clarity). Atom colours: gray – carbon; blue – nitrogen; green – fluorine; brown – zinc; white – hydrogen. | |
 |
| | Fig. 5 Two/three dimensional array of molecules, (a) 1a forming through F⋯F, H⋯H, C⋯C and C⋯H interactions viewed down ‘a’ axis; (b) 2a forming through C⋯F and F⋯H interactions viewed down ‘b’ axis; (c) 3c forming through N⋯H, C⋯H, C⋯N and N⋯N interactions viewed down ‘a’ axis; (d) 3c forming cavity filled with solvent THF molecules viewed down ‘bc’ plane; (e) 6a forming through F⋯F, F⋯H and C⋯H interactions viewed down ‘bc’ plane; (f) 6c viewed down ‘b’ axis. | |
In contrast, the crystal structure of 3c is forming an interesting network of molecules through Zn–N coordination showing a large number of channels present in two directions (Fig. 5d) that are occupied by the solvent (THF) molecules. Notably, the dimension of the cavity in the existing system is smaller compared to the zinc porphyrin–MOF reported with the tetratopic ligand which has larger dimensions present in all the three directions.38 The average Zn–Npyrrole and Zn–Npyridine distances are found to be 2.059 (3) and 2.401 (11) Å. This array of porphyrin molecules is exhibiting an orthogonal fashion of arrangements between them.
Fig. 5c and d represents the molecular crystal packing of 3c in the absence and presence of solvent THF molecules at the cavities. Interestingly, the solvated THF molecule present inside the cavities of 3c was not involved in any kind of intermolecular interactions with the porphyrin molecule. The packing diagram of 6a and 6c are also shown in Fig. 5e and f in which the former forms a one dimensional array of molecules through many intermolecular interactions listed in Table 4. Interestingly, the intermolecular interactions in 6c are mainly through the solvated methanol molecules and trifluoromethanesulfonate anions present in the crystalline lattice and also partly through F⋯H interactions. Both zinc(II) centre in 3c and 6c is hexa-coordinated and exhibits nearly planar geometry and the mean plane deviation of core atoms is found to be ±0.2389 Å and ±0.0497 Å respectively. The pentafluorophenyl ring and 4′-pyridyl/4′-N-methylpyridyl ring make dihedral angles of 66.99°, 67.66° for 3c and 87.69°, 57.03° for 6c with the porphyrin mean plane.
Table 4 Distances (in Å) for different types of interactions in porphyrins, 1a, 2a, 3c, 6a and 6c
| Interactionsa |
1ab |
2ab |
3cb |
6ab |
6cb |
| Different types. Value in parenthesis gives the number of interactions present in the molecule. |
| (pyrrole)C–H⋯C(pyrrole) |
2.841, 2.885 (8) |
— |
— |
2.870 (4) |
— |
| (ph)C–H⋯O(sol) |
— |
— |
— |
— |
2.451 (2) |
| (pyrrole)C–H⋯O(sol) |
— |
— |
— |
2.670 (4) |
— |
| (ph)C–H⋯O(OTf) |
— |
— |
— |
— |
2.370–2.641 (6) |
| (N-methyl)C–H⋯O(OTf) |
— |
— |
— |
— |
2.611 (2) |
| (pyrrole)C–H⋯F(ph) |
2.539 (4) |
2.519 (4) |
— |
— |
2.448 (2) |
| (ph)C–H⋯F(ph) |
— |
2.613, 2.653 (8) |
— |
2.443 (4) |
2.600 (2) |
| (N-methyl)C–H⋯F(ph) |
— |
— |
— |
2.513 (4) |
— |
| (sol)C–H⋯F(ph) |
— |
— |
— |
— |
2.639 (2) |
| (ph)C⋯O(sol) |
— |
— |
— |
— |
3.064 (2) |
| (ph)C⋯O(OTf) |
— |
— |
— |
— |
3.044–3.149 (6) |
| (ph)C⋯F(ph) |
— |
3.01, 3.047 (8) |
— |
— |
3.005, 3.065 (4) |
| (ph)C⋯F(OTf) |
— |
— |
— |
— |
3.120–3.150 (6) |
| (sol)O⋯O(OTf) |
— |
— |
— |
— |
2.799 (2) |
| F⋯F |
2.919 (4) |
— |
— |
2.802 (4) |
— |
| (sol)O–H⋯O(OTf) |
— |
— |
— |
— |
2.008 (2) |
| (sol)O–H⋯S(OTf) |
— |
— |
— |
— |
2.935 (2) |
| (ph)C⋯C(pyrrole) |
3.332 (4) |
— |
— |
— |
— |
| (ph)C–F⋯C(pyrrole) |
3.137 (4) |
— |
— |
— |
— |
| (ph)C–H⋯H(ph) |
2.255 (2) |
— |
— |
— |
— |
| (ph)C⋯C(ph) |
— |
3.208 (2) |
— |
— |
— |
| (ph)C–H⋯N(pyrrole) |
— |
— |
2.682, 2.726 (8) |
— |
— |
| (ph)C⋯N(pyrrole) |
— |
— |
3.219 (4) |
— |
— |
| (ph)N⋯N(pyrrole) |
— |
— |
3.054, 3.068 (4) |
— |
— |
| (ph)C–H⋯C(pyrrole) |
— |
— |
2.870, 2.873 (8) |
— |
— |
It is also noted that 6c is prevented from the Zn⋯N(pyridyl) and N(pyridyl)⋯C interactions because of the N-methylation on the nitrogen of the pyridyl ring. As seen earlier, there are no direct π–π as well as intramolecular interactions in 1a, 2a, 3c, 6a and 6c. And, their crystal packing consists of a number of intermolecular interactions involving carbon, fluorine, hydrogen, oxygen, nitrogen and sulphur, viz., (ph/pyrrole/sol)C–H⋯F, (ph)C–H⋯C(pyrrole), (ph)C–H⋯O(sol/OTf), (ph)C⋯O(sol/OTf), (ph)C⋯F(sol/OTf), (sol)O⋯O(OTf), (sol)O–H⋯O(OTf), (sol)O–H⋯S(OTf), F⋯F, etc. (Table 4).
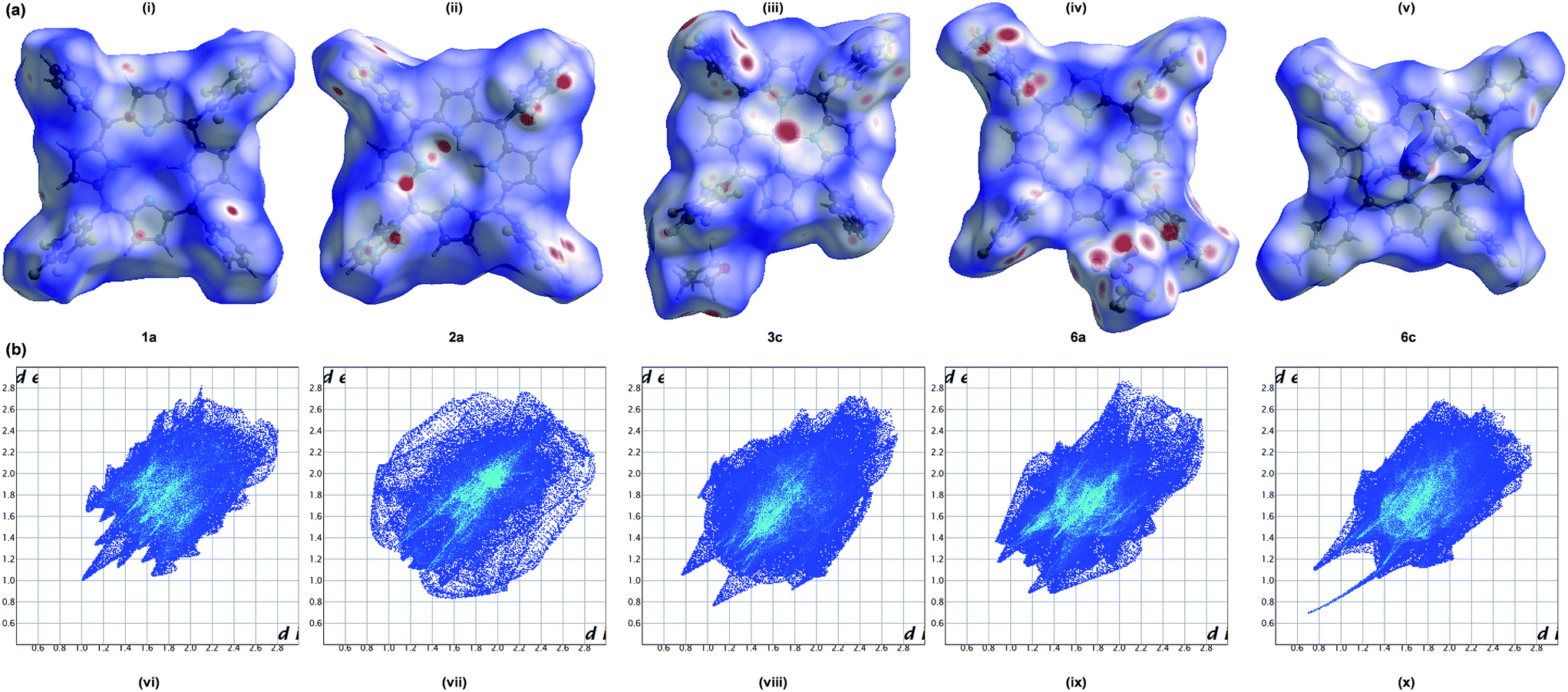
The weak intermolecular interactions present in porphyrins 1a, 2a, 3c, 6a and 6c were analyzed and quantified through Hirshfeld surface analysis using Crystal Explorer 3.1 (Fig. 6). The close contacts can be visualized by colour-coding short or long contacts, the colour and its intensity indicating the relative strength of the interactions. The molecular HSs generated for the porphyrins have different shapes reflecting different kind of crystal packing modes and is further evident from the geometrical analysis (Table 4). The HSs mapped with dnorm highlights intense red spots for H⋯H, C⋯H, Zn⋯N, O⋯H and O⋯S/H close contacts respectively for 1a, 2a, 3c, 6a and 6c. The interactions involving fluorine such as F⋯H/C/F/O features medium/less intense red spots indicate that those interactions are weaker and longer compared to our earlier reports on tetra substituted fluorinated porphyrins.37,39 The breakdown of 2D fingerprint plots (FPs) of the HSs for porphyrins under study reveals that the interactions involving fluorine are 49–58% which demonstrates the influence of halogen atoms on the intermolecular interactions which in turn stabilizes the crystal packing.
 |
| | Fig. 6 (a) Hirshfeld surfaces mapped with dnorm ranging from −0.15 Å (red) to 1.76 Å (blue) and (b) 2D fingerprint plots, (i and vi) 1a; (ii and vii) 2a; (iii and viii) 3c; (iv and ix) 6a; (v and x) 6c. | |
DNA binding studies
Electronic spectral titrations. To investigate the interaction of cationic porphyrins to DNA, the UV-Vis titrations were performed by fixing porphyrin concentration at 3 μM while varying the DNA concentration (Fig. 7). All the cationic porphyrins showed different extent of hypochromism (37–64%) as well as bathochromic response (1–5 nm) at the Soret band which indicates that these porphyrins bind to DNA. It is well known that the porphyrins upon binding with DNA through intercalation results in hypochromism (up to 40%) with red shift (>10 nm) whereas a low hypochromism and small red shift is the signature of outside binding without self-stacking while a large hypochromism with a small red shift is indicative of outside binding with self-stacking.24a The changes in the absorption spectra of porphyrins 4–6 in the absence and presence of CT–DNA were presented in Table 5.
 |
| | Fig. 7 Absorption spectra of pH 7.2 buffer solutions of (a) 4a and (b) 5c in the presence of increasing amounts of CT DNA. [Porphyrin] = 3 μM, [DNA] = 0–3 μM. Inset: plot of [DNA]/(Δε) vs. [DNA]. | |
Table 5 UV-Vis spectral titration parameters for porphyrins, 4–6 with CT–DNA
| Porphyrin |
Δλ (nm) |
Hypochromicity, H (%) |
Kb (M−1) |
| DP-2/3/4-NMPP correspond to 5,15-di(phenyl)-10,20-bis(2′-3′-4′-N-methylpyridyl)porphyrins which are taken from ref: Bioorg. Med. Chem., 2006, 14, 2956–2965. |
| DP-2-NMPPa |
0.0 |
16.20 |
2.50 × 104 |
| DP-3-NMPPa |
0.0 |
39.70 |
8.39 × 104 |
| DP-4-NMPPa |
7.0 |
15.70 |
4.35 × 105 |
| 4a |
3.5 |
58.42 |
2.96 × 105 |
| 4b |
4.5 |
60.10 |
8.47 × 105 |
| 4c |
1.0 |
52.95 |
7.02 × 105 |
| 5a |
5.0 |
61.87 |
1.83 × 106 |
| 5b |
2.0 |
43.85 |
1.50 × 106 |
| 5c |
4.5 |
64.02 |
1.24 × 106 |
| 6a |
4.0 |
43.35 |
1.38 × 106 |
| 6b |
2.5 |
45.30 |
2.00 × 106 |
| 6c |
4.5 |
37.20 |
1.75 × 106 |
In order to further quantify the binding affinities of porphyrins to DNA, the intrinsic binding constant, Kb has been determined based on the absorbance titration data using the following equation:40
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/Kb(εb − εf) |
where
εa,
εf,
εb correspond to
Aobs/[porphyrin], the extinction coefficient for the free porphyrin, and the extinction coefficient for the porphyrin in the fully bound form, respectively. In the plot of [DNA]/(
εa −
εf)
versus [DNA],
Kb is given by the ratio of slope to intercept. The binding constants obtained for the porphyrins
4–6 are in the range of 2.96 × 10
5 to 2.00 × 10
6 M
−1. Interestingly, the
Kb values obtained are higher than the reported non-fluorinated cationic pyridinium porphyrins
41 which discloses that the pentafluorophenyl groups play an essential role to control DNA interaction abilities. Moreover, the binding constant of
6a–6c are almost tenfold higher than the reported Ru(
II) analog of
trans-porphyrin (1.1 × 10
5) that contain both 4-pyridyl as well as fluoro substituents at its periphery.
42 The results clearly signal that among the three sets of porphyrins (
4–6), the binding affinity of 3-/4-
N-methylpyridyl derivatives are higher compared to 2-
N-methylpyridyl derivatives presumably due to the more effective electrostatic interaction between the positively charged porphyrin and the phosphate moiety of nucleotide in the former than the later.
43
Emission studies. The freebase and zinc(II) porphyrins emit luminescence in Tris buffer at pH 7.2. The 2-N-methylpyridyl and 3-N-methylpyridyl freebase porphyrins exhibit two emission bands around 636 nm and 699 nm respectively whereas the corresponding Zn(II) derivatives show two emission bands around 600 and 650 nm respectively. The 4-N-methylpyridyl derivatives shows red shifted emission bands compared to the 2-N-methylpyridyl and 3-N-methylpyridyl derivatives at 648 and 703 nm respectively for the free ligand and 623 and 652 nm for the Zn(II) complex. With the incremental addition of DNA, there is a decrease in fluorescence intensity without much wavelength shift which can be attributed to self-stacking of the porphyrin molecules along the DNA surface (Fig. 8).25
 |
| | Fig. 8 Fluorescence emission spectra of porphyrins, (a) 4a and (b) 6c with increasing concentration of CT DNA. [Porphyrin] = 3 μM, [DNA] = 0–1 μM. λex = 480 nm. | |
Fluorescence quenching experiments were carried out by adding the porphyrins to CT DNA pre-treated with ethidium bromide (EB). EB is an efficient DNA intercalator that emits strong fluorescence upon binding to DNA.44 When EB is replaced or excluded from the interior hydrophobic environments of the DNA double helix by the third molecule, the fluorescence of the EB–DNA system is quenched. Notably, all the porphyrins exhibit remarkable decrease in intensity of fluorescence which indicates that the porphyrins were able to push the intercalated EB out of the nucleotide cavities. The quantitative information of the fluorescence quenching by porphyrins were obtained using the following Stern–Volmer equation:45
| F0/F = 1 + KSV[Q] = 1 + Kqτ0[Q] |
where,
F0 and
F represent the steady-state fluorescence intensity in the absence and presence of the quencher (porphyrin). [Q] is the quencher concentration,
KSV is the Stern–Volmer quenching rate constant which is obtained by the ratio of slope to the intercept of the diagram
F0/
F vs. [porphyrin]. Subsequently, the approximate quenching constant (
Kq) can be calculated from the equation,
KSV =
Kqτ0. The lifetime of the fluorophore (CT–DNA–EB) is
τ0 (19.2 ns) in the absence of the quencher. The value of
Kq is in the order of 10
12 M
−1 S
−1 which is much higher than the maximum diffusion collisional quenching rate of various quenchers with biomacromolecules
46,47 (≈2.0 × 10
10 M
−1 S
−1) (
Table 6), showing the existence of a static quenching mechanism rather than dynamic collision. Since, ethidium bromide is a very strong intercalator, the formation of non-fluorescent (EB–DNA)–porphyrin adduct could be the reason for the static quenching (
Fig. 9).
Table 6 The Stern–Volmer quenching parameters for DNA–ethidium bromide by porphyrins in Tris–buffer at pH = 7.0
| Porphyrin |
4a |
4b |
4c |
5a |
5b |
5c |
6a |
6b |
6c |
| KSV (M−1), 104 |
5.31 |
8.25 |
2.18 |
5.73 |
7.33 |
4.58 |
6.58 |
9.63 |
5.87 |
| Kq (M−1 S−1), 1012 |
2.76 |
4.30 |
1.14 |
2.98 |
3.82 |
2.39 |
3.43 |
5.02 |
3.06 |
 |
| | Fig. 9 Emission spectra of EB bound to DNA with increasing concentration of porphyrins (a) 4a (λex = 411 nm) and (b) 5a (λex = 412 nm) [DNA] = 12 μM, [EB] = 7.5 μM, [porphyrin] = 0–12 μM. Inset: fluorescence quenching curve of EB bound DNA by the porphyrins. | |
The fluorescence quenching efficiency is also calculated from the fluorescence quenching curves and it is found that the highly efficient 3-pyridyl/4-pyridyl derivatives quench the fluorescence around 50–56%.
DNA photocleavage studies. The porphyrins, 4–6 in DMF–Tris buffer containing NaCl (pH = 7.2) and aqueous solutions of circular plasmid DNA (pBR 322) were irradiated using a 15 W transilluminator at 365 nm for 60 min. The irradiated samples were subjected to gel electrophoresis in order to determine the photocleavage ability of the porphyrins.Fig. 10 illustrates the concentration dependent gel electrophoresis experiment for complexes 4a–4c in dark (left) as well as light (right), where lane 1 represents DNA in the absence of porphyrin. Fig. 11 elucidates the photocleavage experiments of porphyrins, 5 and 6. The results indicate that the porphyrins show better activity under light compared to the dark condition. Unlike the reported48 water soluble porphyrin Zn–TTHPP, our zinc(II) derivatives show better photo induced cleavage ability. Moreover, the free ligands also show efficient DNA damage than that of copper(II) counter parts.
 |
| | Fig. 10 Gel electrophoresis of pBR 322 DNA in the absence (lane 1) and presence (lanes 2–7) of porphyrins 4a, 4b and 4c at the dark (a) and after irradiation (b). [DNA] = 0.2 μg; [porphyrin] = 5 μM for lanes 2, 3 and 4. [Porphyrin] = 10 μM for lanes 5, 6 and 7. Photo irradiations were conducted using a UV illuminator (365 nm, 15 W) for 60 min. | |
 |
| | Fig. 11 Gel electrophoresis of pBR 322 DNA in the absence (lane 1) and presence (lanes 2–7) of porphyrins 5a, 5b and 5c (a) and 6a, 6b and 6c (b) after irradiation. [DNA] = 0.2 μg; [porphyrin] = 5 μM for lanes 2, 3 and 4. [Porphyrin] = 10 μM for lanes 5, 6 and 7. Photo irradiations were conducted by using a UV illuminator (365 nm, 15 W) for 60 min. | |
In order to recognize the DNA photocleavage mechanism of porphyrins, we examined the influence of scavengers such as NaN3 and DMSO.49 Photocleavage experiments with NaN3 (singlet oxygen quencher) was carried out and the cleavage of plasmid DNA was inhibited whereas in presence of DMSO, a hydroxyl radical scavenger, the degree of cleavage is almost unaltered. This indicates that not the hydroxyl radical but the singlet oxygen is involved in the DNA photocleavage mechanism (Fig. 12).
 |
| | Fig. 12 Effect of scavengers on the cleavage of DNA in presence of porphyrins: lane 1: DNA control; lane 2: DNA + 4a; lane 3: DNA + 4a + NaN3 (50 mM); lane 4: DNA + 4a + DMSO (50 mM); lane 5: DNA + 4c; lane 6: DNA + 4c + NaN3 (50 mM); lane 7: DNA + 4c + DMSO (50 mM). | |
Measurement of singlet oxygen species. The singlet oxygen involved DNA cleavage can be further visualized using 1,3-diphenylisobenzofuran (DPBF) which is an singlet oxygen quencher (Fig. 13).28–30 The relationship between the change in absorbance by DPBF and irradiation time reflects the 1O2 yield of the compounds. The absorbance of DPBF at 410 nm decreased in the presence of porphyrin with increasing irradiation time and from the slope of the line, the relative rate of 1O2 generation was compared. From the slope values, it is observed that the singlet oxygen yield of freebase porphyrins and its zinc(II) derivatives are higher and the values are comparable with the reported tricationic zinc(II) porphyrin29 whereas it is very high compared to the N-confused porphyrin.30 The zinc(II) porphyrin 5c showed a very high singlet-oxygen quantum yield of 0.93 compared with 0.14 for its copper(II) analogue 5b in DMSO. The measured luminescence lifetimes are also comparable with the reported cationic porphyrins.29 (Table S1†) Notably, the results are in good agreement with that of gel electrophoresis experiments.
 |
| | Fig. 13 Decomposition of DPBF by compounds, (a) 4a–4c; (b) 5a–5c and (c) 6a–6c. Porphyrin (0.5 μM) and DPBF (50 μM) were irradiated in DMSO. | |
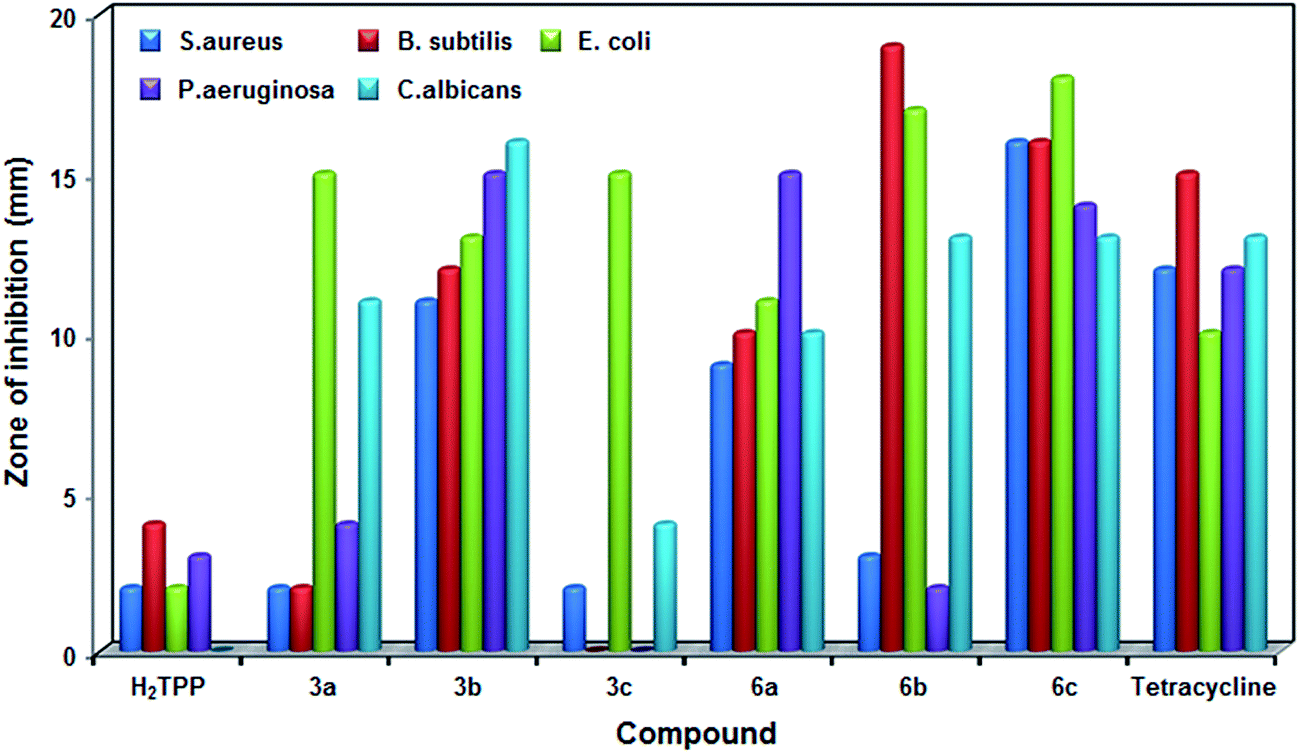
Antimicrobial studies. The antimicrobial activity of trans-4-pyridyl (3a–3c) and trans-4-N-methylpyridyl derivatives (6a–6c) along with H2TPP and standard tetracycline were performed using well diffusion method at three different concentrations (40–160 μM) against the MTCC cultures. The results reveal that the fluorinated analogues showed better activity compared to the non-fluorinated analogue, H2TPP. The growth inhibition observed in zone inhibition assay of metal ions are negligible compared to the porphyrins. It is also observed that all the porphyrins showed maximum zone of inhibition at maximum concentration with MIC values in the range of 6.7 to 16.6 mM against the investigated strains (Fig. 14 & 15). At the concentration of 63 μM, porphyrin 6b showed maximum zone of inhibition of 19 mm and 17 mm against B. subtilis and E. coli respectively, 6c showed inhibition zone of 18 mm against E. coli. The porphyrin 3c did not show any activity against B. subtilis and P. aeruginosa. In the case of the fungal strain C. albicans, maximum activity is found for porphyrin 3b followed by 6b and 6c, no activity is found for H2TPP and very less activity of 4 mm is found for 3c (Fig. 14). Among the samples used, highest MIC was 16.6 mM for sample 3a against C. albicans followed by 16.1 mM for 3a against B. subtilis. Lowest MIC was recorded for 6b (6.7 mM) against B. subtilis followed by porphyrin 6a against E. coli (6.8 mM). Interestingly, the trans-porphyrins under study exhibit better activity towards the fungi C. albicans compared to the reported electron deficient fluorinated metalloporphyrins containing trifluoromethylphenyl groups.50 The porphyrin molecules are likely to alter the structure of cell membrane owing to their affinity to biological organisms which might be the cause for its biological activity.51
 |
| | Fig. 14 Antimicrobial studies of the trans-4-pyridyl and trans-dicationic 4-pyridinium porphyrins along with H2TPP and standard tetracycline (% of error: ±5). | |
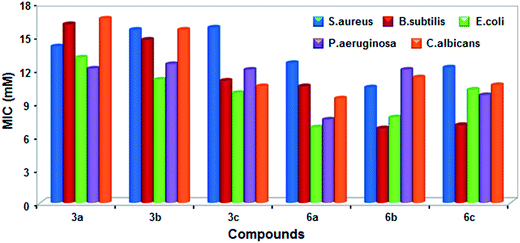 |
| | Fig. 15 The minimum inhibition concentration of trans-4-pyridyl and trans-dicationic 4-pyridinium porphyrins. | |
Cytotoxicity of porphyrins under antimicrobial study in normal Vero cell lines was performed by means of a colorimetric assay (MTT assay). The results showed no variation in the morphological features of the cells at 100 μg mL−1 concentration. Thus, the compounds were found to be safe while treated against normal cells and showed very less toxicity (6–10%) on the normal Vero cells (Fig. S9†).
Conclusions
A series of trans-pyridyl porphyrins, trans-dicationic pyridinium porphyrins and its metal derivatives [Cu(II) and Zn(II)] were synthesized, characterized using conventional spectroscopic methods, electrochemical studies as well as by single crystal X-ray crystallographic analysis. The title compounds exhibited red shifted absorption spectra than the simple pyridyl porphyrins. The reduction potentials of trans-pyridyl porphyrins are more positive than those of MTPPs. X-ray crystal structures of 1a, 2a, 3c, 6a and 6c indicates that the interactions involving fluorine contributes considerably to the crystal packing which were further confirmed by computational Hirshfeld surface analysis. The dicationic porphyrins were further explored for its DNA interaction abilities and antimicrobial activities. Studies reveal that among the studied porphyrins (4–6), the 3-/4-pyridinium derivatives were found to be efficient groove binders with high intrinsic binding constant (Kb) values compared to its corresponding non-fluorinated analogues reported earlier. Hence, the introduction of fluorine into the phenyl rings as well as the position of positive charges on the N-methylpyridinium group of porphyrin greatly influences their interaction with DNA. The photocleavage experiments disclose that the porphyrins employ 1O2-mediated mechanism in cleaving DNA and the freebase, zinc(II) derivatives show better photoinduced cleavage ability compared to its copper(II) analogues. Among the porphyrins studied, the zinc(II) complex, 6c shows the most promise as a photosensitizer for PDT. The dicationic porphyrins also show significant antimicrobial activities than those of non-fluorinated analogues. Thus, the results obtained from the present work would be further explored in medicinal chemistry as new potent therapeutic agents.
Acknowledgements
SS (SR/WOS-A/CS-146/2011) thank DST, New Delhi for the financial support. We would like to thank Dr Babu Varghese, SAIF, IIT Madras for the single crystal data collection, structure solution and refinement. We also thank Dr Leeba Balan, University of Madras, Chennai, Tamilnadu for helping us to acquire the antimicrobial studies.
References
- R. J. Fiel, J. Biomol. Struct. Dyn., 1989, 6, 1259–1274 CAS.
- S. Swavey and M. Tran, Porphyrin and Phthalocyanine Photosensitizers as PDT Agents: A New Modality for the Treatment of Melanoma, Recent Advances in the Biology, Therapy and Management of Melanoma, ed. L. Davids, InTech, 2013, ISBN: 978-953-51-0976-1, DOI:10.5772/54940.
- The Porphyrin Handbook, ed. K. Kadish, K. Smith and R. Guilard, Academic Press, New York, 2000, vol. 1–20 Search PubMed.
- E. D. Sternberg and D. Dolphin, Tetrahedron, 1998, 54, 4151–4202 CrossRef CAS.
- R. J. Fiel, J. C. Howard, E. H. Mark and N. Datta Gupta, Nucleic Acids Res., 1979, 6, 3093–3118 CrossRef CAS PubMed.
- C. Wei, G. Jia, J. Yuan, Z. Feng and C. Li, Biochemistry, 2006, 45, 6681–6691 CrossRef CAS PubMed.
- V. G. Barkhudaryan, G. V. Ananyan, Y. B. Dalyan and S. G. Haroutiunian, J. Porphyrins Phthalocyanines, 2014, 18, 594–599 CrossRef CAS.
- B. Armitage, Chem. Rev., 1998, 98, 1171–1200 CrossRef CAS PubMed , and references therein.
- S. Cogoi and L. E. Xodo, Chem. Commun., 2010, 46, 7364–7366 RSC.
- C. Shi and F. C. Anson, J. Am. Chem. Soc., 1991, 113, 9564–9570 CrossRef CAS.
- N. A. Stephenson and A. T. Bell, Inorg. Chem., 2007, 46, 2278–2285 CrossRef CAS PubMed.
- T. Goslinski and J. Piskorz, J. Photochem. Photobiol., C, 2011, 12, 304–321 CrossRef CAS.
- M. Boccalon, E. Lengo and P. Tecilla, Org. Biomol. Chem., 2013, 11, 4056–4067 CAS.
- M. Vinodu and I. Goldberg, CrystEngComm, 2005, 7, 133–138 RSC.
- X. Chen, L. Hui, D. A. Foster and C. M. Drain, Biochemistry, 2004, 43, 10918–10929 CrossRef CAS PubMed.
- S. Rani-Beeram, K. Meyer, A. McCrate, Y. Hong, M. Nielsen and S. Swavey, Inorg. Chem., 2008, 47, 11278–11283 CrossRef CAS PubMed.
- S. P. de Visser and W. Nam, High-valent iron-oxo porphyrins in oxygenation reactions, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co, New Jersey, 2010, ch. 44, pp. 85–140, and references therein Search PubMed.
- D. D. Perrin and W. L. F. Armarego, Purification of Organic Solvents, Pergamon Press, Oxford, 1988 Search PubMed.
- B. J. Littler, M. A. Miller, C. H. Hung, R. W. Wagner, D. F. O'Shea, P. D. Boyle and J. S. Lindsey, J. Org. Chem., 1999, 64, 1391–1396 CrossRef CAS.
- A. G. Altomare, G. Cascarano, C. Giacovazzo and A. Gualardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, SHELXL97, University of Goettingen, Goettingen, Germany, 1997 Search PubMed.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, Crystal Explorer 3.1 2013, University of Western Australia, Crawley, Western Australia, 2005-2013, http://www.hirshfeldsurface.net/CrystalExplorer Search PubMed.
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
-
(a) R. F. Pasternack and E. J. Gibbs, Porphyrin and Metalloporphyrin Interactions with Nucleic acids, Metal ions in Biological Systems, Marcel Dekker Inc, New York, 1996 Search PubMed;
(b) Z. Assefa, A. Vantieghem, Q. Deelereq, P. Vandenabeele, J. R. Vandenheede, W. Merlevede, P. deWitte and P. Agostinis, J. Biol. Chem., 1999, 274, 8788–8796 CrossRef CAS PubMed.
- P. Zhao, J.-W. Huang, W.-J. Mei, J. He and L.-N. Ji, Spectrochim. Acta, Part A, 2010, 75, 1108–1114 CrossRef PubMed.
- T. Sarwar, M. A. Husain, S. U. Rehman, H. M. Ishqi and M. Tabish, Mol. BioSyst., 2015, 11, 522–531 RSC.
-
(a) B. Armitage, Chem. Rev., 1998, 98, 1171–1200 CrossRef CAS PubMed;
(b) P. Sweigert, Z. Xu, Y. Hong and S. Swavey, Dalton Trans., 2012, 41, 5201–5208 RSC;
(c) N. Chitrapriya, J. Park, W. Wang, H. Lee and S. K. Kim, Metallomics, 2012, 4, 417–421 RSC.
- R. Soman, D. Raghav, S. Sujatha, K. Rathinasamy and C. Arunkumar, RSC Adv., 2015, 5, 61103–61117 RSC.
- P. M. Antoni, A. Naik, I. Albert, R. Rubbiani, S. Gupta, P. Ruiz-Sanchez, P. Munikorn, J. M. Mateos, V. Luginbuehl, P. Thamyongkit, U. Ziegler, G. Gasser, G. Jeschke and B. Spingler, Chem.–Eur. J., 2015, 21, 1179–1183 CrossRef CAS PubMed.
- P. Thomas, P. S. S. Babu, S. Asha Nair, S. Ramakrishnan, D. Ramaiah, T. K. Chandrashekar, A. Srinivasan and M. R. Pillai, J. Med. Chem., 2012, 55, 5110–5120 CrossRef PubMed.
- D. Greenwood, R. Snack and J. Peurtherer, Medical Microbiology, A Guide to Microbial Infections, Pathogenesis, Immunity, Laboratory Diagnosis and Control, 15th edn, 1997 Search PubMed.
- B. A. Wariso and O. Ebong, Int. J. Pharm. Sci. Drug Res., 1996, 12, 65–68 Search PubMed.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52, 827–836 CrossRef CAS.
-
(a) P. Bhyrappa and V. Krishnan, Inorg. Chem., 1991, 30, 239–245 CrossRef CAS;
(b) P. Bhyrappa, S. R. Wilson and K. S. Suslick, J. Am. Chem. Soc., 1997, 119, 8492–8502 CrossRef CAS;
(c) W. Wu, W. Wu, S. Ji, H. Guo, X. Wang and J. Zhao, Dyes Pigm., 2011, 89, 199–211 CrossRef CAS;
(d) X. Wang, X. Chen, Z. Xie and X. Wang, Angew. Chem., Int. Ed., 2008, 47, 7450–7453 CrossRef CAS PubMed.
- K. M. Kadish, E. Van Caemelbecke and G. Royal, Electrochemistry of Metalloporphyrins in Nonaqueous Media, in The Porphyrin Handbook, ed. K. Kadish, K. Smith and R. Guilard, Academic Press, New York, 2000, vol. 8, ch-55 Search PubMed.
- E. Z. Moreira, A. D. Q. Ferreira, C. R. Neria, S. Mukhopadhyay, S. Dovidauskas, S. Nikolaou and Y. Iamamoto, J. Porphyrins Phthalocyanines, 2010, 14, 975–984 CrossRef CAS.
- R. Soman, S. Sujatha, S. De, V. C. Rojisha, P. Parameswaran, B. Varghese and C. Arunkumar, Eur. J. Inorg. Chem., 2014, 2653–2662 CrossRef CAS.
- A. M. Shultz, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 4204–4205 CrossRef CAS PubMed.
- R. Soman, S. Sujatha and C. Arunkumar, J. Fluorine Chem., 2014, 163, 16–22 CrossRef CAS.
- R. F. Pasternack, E. J. Gibbs and J. J. Villafranca, Biochemistry, 1983, 22, 2406–2414 CrossRef CAS PubMed.
-
(a) S. Wu, Z. Li, L. Ren, B. Chen, F. Liang, X. Zhou, T. Jia and X. Cao, Bioorg. Med. Chem., 2006, 14, 2956–2965 CrossRef CAS PubMed;
(b) D. Kumar, B. Mishra, K. P. Chandrashekar, S. B. Khandagale, M. P. Tantak, A. Kumar, K. Akamatsu, E. Kusaka, K. Tanabe and T. Ito, RSC Adv., 2015, 5, 53618–53622 RSC.
- M. Narra, P. Elliott and S. Swavey, Inorg. Chim. Acta, 2006, 359, 2256–2262 CrossRef CAS.
- G. Pratviel, Coord. Chem. Rev., 2016, 308, 460–477 CrossRef CAS.
- F. J. Meyeralmes and D. Porschke, Biochemistry, 1993, 32, 4246–4253 CrossRef CAS.
- J. K. Lakowicz, Principles of Fluorescence Spectroscopy Plenum Press, New York, 2nd edn, 1999 Search PubMed.
- Biophysical and Biochemical Aspects of Fluorescence Spectroscopy, ed. T. G. Dewey, Plenum Press, New York, 1991 Search PubMed.
- Z. Mandegani, Z. Asadi, M. Asadi, H. R. Karbalaei-Heidari and B. Rastegari, Dalton Trans., 2016, 45, 6592–6611 RSC.
- S. C. Karunakaran, D. Ramaiah, I. Schulz and B. Epe, Photochem. Photobiol., 2013, 89, 1100–1105 CrossRef CAS PubMed.
- M. A. Sari, J. P. Battioni, D. Dupre, D. Mansuy and J. B. Le pecq, Biochemistry, 1999, 29, 4205–4215 CrossRef.
- F. R. Kooriyaden, S. Sujatha and C. Arunkumar, Polyhedron, 2015, 97, 66–74 CrossRef CAS.
- A. Hou, Z. Xue, Y. Liu, S. Qu and W. Wang, Chem. Biodiversity, 2007, 4, 1492–1500 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data of the porphyrins, 1a, 2a, 3c, 6a and 6c. CCDC 1434316, 1443805, 1021573, 1058021 and 1041762. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra09148b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.