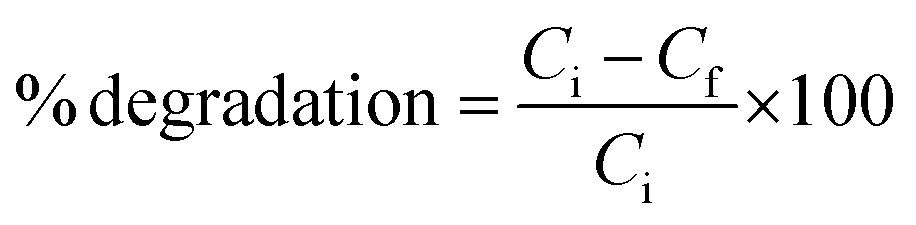DOI:
10.1039/C6RA09110E
(Paper)
RSC Adv., 2016,
6, 56790-56799
Template-free syntheses of hierarchical PbS microstructures using a new sulphur source and their time-dependent morphological evolution and photocatalytic properties†
Received
8th April 2016
, Accepted 6th June 2016
First published on 7th June 2016
Abstract
Template-free lead sulphide (PbS) microstructures composed of nanocrystals exhibiting time-dependent morphological evolution from cubes to dendrites (S1–S5) were synthesised by a one-pot solvothermal route using dibenzyl disulphide (DBDS = (C7H7S)2) as a new temperature controlled in situ source of S2− ions. Use of mercaptoethanol (MCE) capping agent resulted in the formation of PbS microstructures (S6–S10) with mainly dendritic morphology as the major phase. Powder X-ray diffraction measurements unveiled the cubic structure of the PbS microstructures. FESEM analyses of the uncapped PbS microstructures (S1–S5) obtained at different time intervals revealed time-dependent morphological evolution from cubic to dendritic structures, whereas the MCE-capped PbS microstructures show the formation of mostly dendritic structures. The possible mechanism of the morphological evolution of PbS microstructures in the presence and absence of MCE capping agent has been discussed. Optical measurements of the samples show a blue-shift in the absorption maxima in comparison to that of bulk PbS and the estimated band gap values are in the range of 3.5–3.8 eV. Photocatalytic investigation of cubic and dendritic microstructures for the degradation of MO revealed a higher catalytic activity of dendrites over cubes. The relatively higher activity of dendrites has been attributed to the higher charge transfer originating due to the branched nature of dendrites. Thus the influence of capping agent on the morphological evolution and the photocatalytic activity of PbS hierarchical structures has been presented.
Introduction
In modern materials chemistry the architectural control of nanocrystals with well defined morphology is important to tune their optical,1 electronic,2 magnetic3 and catalytic4 properties. It is well-known that the morphology of nanoparticles has a profound influence on their properties. In this context, shape controlled syntheses of semiconductor nanoparticles has attracted intensive interest. Moreover the ability to understand and predict the architecture of nanoscale materials is still limited. If the shape-guiding processes in these building blocks are understood, it could be possible to program the system to yield the final products with desired morphology and properties.
PbS (galena) is the major source of lead and is an important IV–VI semiconductor with a narrow band gap energy of 0.41 eV with a large excitation Bohr radius of 18 nm and has potential applications in near-IR communications and switches.5 Various methods have been developed for the syntheses of PbS submicro-/nanocrystals by employing solvothermal methods,6 chemical vapour deposition (CVD),7 ultrasonic methods8 etc. Different morphologies of PbS nanocrystals have been prepared such as nanocubes,9 nanoflowers,10 stars,11 nanowires,12 matchstick13 and dendrites.14 However, a mechanistic study of the formation of these structures is elusive. In general, the formation of these morphologies are assisted by templates/capping agents.15 On the other hand, detailed investigations on morphological evolution of PbS nanocrystals formed in the absence of templates/capping agents are rare.16 We have been attempting to synthesis template-free semiconductor nanostructures using new temperature controlled in situ source of S2− ions, with controlled morphologies.17 In continuation of our efforts, herein we report for the first time the synthesis of PbS microstructures (S1–S5) using DBDS as a temperature controlled in situ source of S2− ion in the absence of a template/capping agent and their time-dependent morphological evolution from cubic (isotropic) to dendritic (anisotropic) structure. Furthermore, to understand the effect of capping agent on the morphology of PbS microstructures, MCE-capped PbS microstructures (S6–S10) have also been prepared. Interestingly, the MCE-capped PbS microstructures exhibit mainly dendritic morphology as opposed to the uncapped PbS. Optical measurements of all the samples S1–S10 showed blue-shift in the UV-vis absorption maxima as compared to that of the bulk PbS. Morphology dependent photocatalytic investigation of both cubic (S2) and dendritic (S7) PbS structures for degradation of methyl orange (MO) dye under UV-light irradiation revealed higher photocatalytic activity of S7 as compared to that of S2. The relatively higher photocatalytic activity of dendrites over cubes has been attributed due to the branched nature of the former which facilitates better charge transfer properties. The development of unique synthesis procedure for PbS microstructures and their detailed time-dependent morphological evolution and the photocatalytic activity with and without MCE-capping agent is presented.
Experimental section
Materials
All the starting materials were commercially available and used as received without further purification. Lead(II) nitrate (Pb(NO3)2) and dibenzyl disulphide (DBDS) were purchased from Sigma-Aldrich Chemical Co. Dimethylformamide (DMF), ethanol (EtOH), methanol (MeOH) were obtained from Merck. Methyl orange (MO) was obtained from Spectrochem Chemical Co.
Physical measurements
The powder X-ray diffraction (XRD) patterns were recorded on a PANalytical's X'PERT PRO diffractometer. Field-emission scanning electron microscopy (FESEM) images were recorded on a Carl Zeiss Ultra 55 FESEM with Energy Dispersive Spectroscopy (EDS) facility. UV-vis spectra were recorded on a UV-2600 Schimadzu corporation spectrophotometer. Fourier-transform infrared (FT-IR) measurements were recorded on Bruker TENSOR-27 spectrometer.
Syntheses of PbS microstructures
0.1 mmol of Pb(NO3)2 was dissolved in 6 ml of dimethylformamide (DMF) and to this, ethanolic solution (3 ml) of DBDS (0.1 mmol) was added. The mixture was stirred for 30 minutes and then taken in a 23 ml PTFE lined acid digestion bomb and heated at 150 °C for different time intervals of 9, 12, 18, 24, 30 h. After being cooled to room temperature the black product was filtered, washed with methanol few times and dried under vacuum and the samples were labeled as S1, S2, S3, S4 and S5 respectively. Similar reaction procedure was used for the synthesis of MCE-capped PbS in the presence of MCE-capping agent and the samples obtained with reaction time 9, 12, 18, 24, 30 h were labeled as S6, S7, S8, S9 and S10 respectively. The syntheses of uncapped PbS was repeated by following the procedure as described above but carried out at temperatures of 100, 120, 130, 180 and 200 °C with time interval of 9 h for all the samples.
Photocatalytic study
The morphology-dependent photocatalytic activity of the as-prepared samples of PbS microstructure showing cubic and dendritic morphology for the photocatalytic degradation of an aqueous solution of MO was carried out as follows. The as-prepared sample of PbS microstructures (S2/S7) (25 mg each) was suspended in a solution of MO (2.0 × 10−5 M) in 40 ml of H2O in a quartz tube at ambient temperature. A 400 W high pressure mercury vapour lamp was used as the UV-vis light source. The lamp was equipped with double-walled quartz jacket for constant water circulation and also as a water filter to remove the heating effects. The distance between the UV light source and the quartz reaction tube was maintained at 15 cm. Before irradiation, the mixture was stirred well in the dark for 30 minutes to establish an adsorption–desorption equilibrium between the PbS microstructures and MO and then the photocatalytic reaction was initiated. The reaction was started after the intensity of the high pressure mercury vapour lamp became stable. At regular intervals of time, aliquots of the solution were withdrawn and the catalyst was separated through centrifugation and the supernatant solution was analysed by UV-vis spectrophotometer. The percentage degradation of the dye was calculated using the following formula:
where, Ci and Cf are the initial and the final concentrations of MO respectively.
Results and discussions
Template-free synthesis of uncapped (S1–S5) and MCE-capped PbS (S6–S10) microstructures with controlled morphologies has been achieved using DBDS as a new temperature controlled in situ source of S2− ions without the use of any template/capping agent (Scheme 1). Interestingly these samples exhibit time-dependent morphological evolution from cubic to dendritic structures. On the contrary, the use of MCE-capping agent resulted in the formation of PbS microstructures S6–S10 with mainly dendritic morphology. The powder XRD pattern of the as-prepared PbS samples is shown in Fig. 1. The diffraction peaks observed at 2θ = 26.06, 30.17, 43.15, 51.07, 53.51, 62.62, 68.97 can be assigned to (111), (200), (220), (311), (222), (400) and (331) planes of the cubic structure of PbS (JCPDS reference card no. 05-0592). No additional diffraction peaks corresponding to either Pb(NO3)2 or other impurities were detected indicating the high purity of the samples. The value of crystallite size (D) calculated using the Scherrer equation (D = 0.9λ/(β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ); where, λ is the wavelength of X-ray (1.5405 Å), β is the value of full width at half maximum (FWHM) and θ is the Bragg's angle) is about 50 nm. Further, the composition of the samples was determined by energy dispersive X-ray spectroscopy (EDS). As shown in Fig. 2, the EDS plots of uncapped PbS microstructures, S5 show peaks due to Pb and S atoms only and the quantitative analysis confirmed the atom ratio of Pb/S is about 1:1. Whereas, the EDS plot of MCE-capped PbS, S6 shows additional peak corresponding to oxygen atom of the MCE-capping agent. Furthermore, FT-IR spectra (Fig. S1, ESI†) of un-capped PbS (S2) shows no stretching bands corresponding to –OH and –SH supporting the capping agent free formation of the samples, S1–S5. Whereas the FT-IR spectra of MCE-capped PbS sample, S7 shows stretching bands due to –OH and –SH functional groups of the MCE-capping agent. Therefore, the above mentioned analysis clearly support the formation of PbS microstructures without (S1–S5) and with MCE-capping agent (S6–S10).
cosθ); where, λ is the wavelength of X-ray (1.5405 Å), β is the value of full width at half maximum (FWHM) and θ is the Bragg's angle) is about 50 nm. Further, the composition of the samples was determined by energy dispersive X-ray spectroscopy (EDS). As shown in Fig. 2, the EDS plots of uncapped PbS microstructures, S5 show peaks due to Pb and S atoms only and the quantitative analysis confirmed the atom ratio of Pb/S is about 1:1. Whereas, the EDS plot of MCE-capped PbS, S6 shows additional peak corresponding to oxygen atom of the MCE-capping agent. Furthermore, FT-IR spectra (Fig. S1, ESI†) of un-capped PbS (S2) shows no stretching bands corresponding to –OH and –SH supporting the capping agent free formation of the samples, S1–S5. Whereas the FT-IR spectra of MCE-capped PbS sample, S7 shows stretching bands due to –OH and –SH functional groups of the MCE-capping agent. Therefore, the above mentioned analysis clearly support the formation of PbS microstructures without (S1–S5) and with MCE-capping agent (S6–S10).
 |
| | Scheme 1 Synthesis scheme for the formation of uncapped cubic and MCE-capped dendritic PbS microstructures. | |
 |
| | Fig. 1 (a) Powder XRD patterns of as-synthesized uncapped PbS microstructures, S1–S5 isolated at different time intervals (b) MCE-capped PbS microstructures S6–S10. | |
 |
| | Fig. 2 EDS spectra of as-synthesized samples of un-capped, S5 and MCE-capped, S6 PbS. | |
The time-dependent morphological evolution of as-synthesized PbS samples at different time intervals, S1–S10 was studied by FESEM analysis. As shown in Fig. 3a and b, FESEM images for the PbS sample isolated at 9 h show the presence of well defined cubic morphology. While the samples isolated with time intervals of 12 (S2), 18 (S3), 24 (S4) and 30 h (S5) show the presence of aggregated cubic, octahedron, star-shaped dendrites and dendritic morphologies respectively, Fig. 3b–i. Thus, the FESEM analyses revealed the time-dependent morphological change from cubes to dendrites enroute to the formation of octahedral structures.
 |
| | Fig. 3 FESEM images of as-synthesized PbS microstructures showing time-dependent morphological evolution (a and b) 9 h S1, (c and d) 12 h S2, (e and f) 18 h S3, (g and h) 24 h S4, (i) 30 h S5. | |
On the other hand, MCE-capped PbS microstructures prepared at different time intervals show dendritic morphology as a major phase along with observation of few octahedrons as the minor phase (Fig. 4–6). The octahedrons can be considered as an intermediate phase before the formation of dendrites. From the above discussion it is clear that the presence of capping agent promotes the formation of dendrites at much faster rate as compared to that of the absence of capping agent.
 |
| | Fig. 4 FESEM images of as-synthesized PbS (S6) structures showing the presence of (a) dendrites (b) magnified structure of dendrites and octahedrons (c) growth pattern of individual dendrites (inset image showing the surface of dendrite) (d) six-arm dendritic structures formed at the end of 9 h. | |
 |
| | Fig. 5 FESEM image of the selected region of MCE-capped PbS microstructures, S6 showing the growth of larger dendrites by merging of smaller octahedral structures. | |
 |
| | Fig. 6 FESEM images of PbS microstructures (a–c) S7, (d–f) S8, (g–i) S9, and (j–l) S10 showing the presence of dendrites and six-arm dendritic structures. | |
Mechanism of evolution of morphology
Time-dependent morphological evolution in the absence of capping agent. The morphological evolution of PbS microstructures can be explained by considering the mechanism as shown in Scheme 2(a). The formation of hierarchical PbS microstructures composed of nanocrystals is determined by the nucleation, and the subsequent growth processes of the crystallites. Furthermore, the growth process depends on the crystal habit associated with the surface energy of the exposed planes of the crystal and its branching process depends upon the diffusion effect which is governed by concentration of the precursors and the experimental conditions. Thus the final morphology of the microstructure depends on the crystallographic plane of the seeds formed during the nucleation process. It is known that different crystallographic planes have different surface energies. The general sequence of surface energies of crystallographic planes is elucidated as {111} < {100} < {110}.18 The intrinsic surface energy of the cubic PbS {111} plane that contain pure Pb or S has been found to be higher than that of the {100} plane which contains mixed Pb/S.19 As explained by Wang and co-workers, the value of R, the ratio between the growth rates of <100> and <111> directions mainly decide the morphology of an fcc nanocrystal.20 It has been found that when the value of R = 1.73, octahedrons were formed by most stable {111} plane, while if R was decreased to 0.58, only cubes were formed by the less stable {100} plane. Thus the formation of hierarchical structures can be explained on the basis of RS/Pb (the ratio of sulphur to metal concentration). Here, since DBDS undergoes the temperature controlled in situ release of S2− ions, there is higher concentration of sulphur in the initial stages of the reaction. A higher concentration of sulphur favors the growth along {111} plane resulting cubic structure of PbS in the initial stages of the reaction when nucleation is dominant as shown in Fig. 3a and b.21 As more of the sulphur is utilized in the formation of the cubes, there is decrease in the value of RS/Pb in the reaction mixture leading to morphological instability and the growth of cube begins at this stage with bulges appearing on the surface of the cube (Fig. 3c–e). Similar observation of formation of bulges due to either temperature or concentration gradient has been reported before.22 In our case, since temperature is kept constant the formation of such bulges can be attributed to the concentration gradient. The morphological instability can be explained on the basis of theorems of Berg and Sunagawa.22,23 According to Berg effect, the growth is faster at the planes which are in contact with the inner solution i.e. the corners grow faster than the faces of the cubes. The cubes that are in contact with the inner solution start agglomerating for the growth to occur as is clearly seen from the Fig. 3c–e. The initial cubic seeds dissolve and are transformed into hollow structures which grow into octahedral structures and then to star-shaped dendrites by growth along <100> direction as can be seen from Fig. 3.24
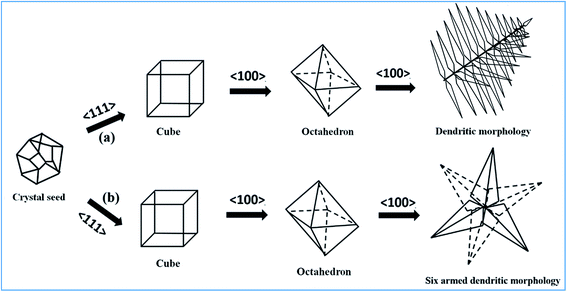 |
| | Scheme 2 (a) Proposed mechanism for the time-dependent morphological evolution of cubes to dendritic morphology of uncapped PbS and (b) formation of six-armed dendritic morphology of MCE-capped PbS. | |
Sunagawa correlated the growth mechanism with the driving force for the formation of such morphologies. The driving force has certain critical value22 and if the driving force is above the critical value for the nucleation to occur the interface morphology loses its stability and the bulges are enhanced. As saturation is reached in the formation of cubes by growth along {111} outcropping occurs along the corners and the growth along {100} is favored to give rise to octahedral and eventually the dendritic structures as seen in case of S5 at the end of 30 h (Fig. 3i). If the driving force is below the critical value then spiral type of growth mechanism is favored. In between the high and low critical values layer by layer two-dimensional growth occurs.
Morphological evolution in the presence of MCE-capping agent. To probe the role of surfactant on the morphological evolution of PbS microstructures we carried out the reaction in the presence of MCE capping agent and the morphological change observed can be explained as shown in Scheme 2b. Mercaptoethanol has –SH functional moiety and is polar in nature. It is known from literature that in a cube, {111} plane is more polar than {100} plane and the –SH group has affinity to bind with more polar {111} plane.25 As discussed above absence of MCE-capping agent yielded cubic structure in the initial stages of the reaction. The presence of MCE-capping agent at this stage will lead to the binding of the –SH moiety to the {111} plane of cube leading to lowering of the surface energy of the plane. Lowering of surface energy hinders the growth along {111} plane and favors along {100} plane resulting in the formation of dendrites as shown in Fig. 4. FESEM images of the MCE-capped PbS sample (S6) isolated at 9 h shows the formation of dendrites as a major phase along with few octahedrons (Fig. 4). The formation of octahedrons can be envisioned through the formation of cubic structures and soon after the cubical seed formed binding of MCE-capping agent results in faster growth along {100} plane yielding dendritic structures enroute to the formation of octahedrons as depicted in Scheme 2b. In support of the above discussed mechanism, Fig. 5 shows the presence of dendrites and octahedrons of different sizes and the growth of larger dendrites by merging of octahedrons on the tips of smaller dendrites. Similarly, FESEM images of the MCE-capped PbS samples S7–S10 isolated at 12, 18, 24 and 30 h, respectively show the presence of dendrites as the major phase as shown in Fig. 6. The growth patterns of dendrites show the formation of six-armed dendritic structures in all samples S7–S10 suggesting that the reaction time has no significant effect on the morphology of MCE-capped PbS as opposed to the un-capped PbS. The formation of these dendrites can be envisioned to proceed through the formation of intermediates of octahedrons as observed in case of sample S6 (Fig. 4b). Thus capping agent play a significant role in fastening the growth process of dendrites by decreasing the energy of {111} plane and facilitating the growth along {100} plane.26
Effect of temperature on the morphological evolution of PbS microstructures. To understand the role of reaction temperature on the morphological evolution of PbS microstructures, the synthesis of PbS was carried out at different temperatures such as, 100, 120, 130, 180 and 200 °C. The reaction carried out at 100 °C did not yield enough compound for the characterization. Whereas, analysis of the PXRD patterns of the samples obtained at 120 (S12), 130 °C (S13) revealed the formation of a new phase other than PbS and starting materials (Fig. S2†) which could be attributed due to the insufficient release of S2− ions at temperatures lower than 150 °C. This supports the fact that the release of S2− ions from DBDS is dependent on the temperature of the reaction. Further, the reactions carried out at 180 (S14), and 200 °C (S15) resulted in the formation of PbS (Fig. S2,† JCPDS reference card no. 05-0592) indicating that at temperatures higher than 150 °C there is efficient release of S2− ions for the formation of PbS. However, the SEM analysis of the samples S14 and S15 showed the growth of agglomerated microstructures (Fig. S3†) which could be ascribed due to faster release of S2− ions. Therefore, from the above discussion it can be concluded that temperature of synthesis has a profound effect on the growth of PbS microstructures, at temperatures lower than 150 °C there is insufficient release of S2− ions hence there is no formation of PbS microstructures. While at temperatures higher than 150 °C there is faster release of S2− ions leading to uncontrolled growth of PbS.
Optical properties
The room temperature solid state UV-vis-IR absorption spectra of as-synthesized PbS microstructures was calculated from Kubelka–Munk function and are shown in the Fig. 7a for S1–S5 and Fig. 7b for S6–S10. The absorption edges of the PbS microstructures, S1–S5 are found to be at 278, 596, 865 nm and for S6–S10 have been found to be at 250, 600, 1240 nm. The blue-shift in the absorption maxima of the samples in comparison to that of bulk PbS (3020 nm) can be attributed due to the position-dependent quantum confinement effect.27,28 For PbS microstructures with crystallite size larger than the exciton Bohr radius (18 nm), the exact assignment of absorption maxima has been quite difficult.29 For such systems it has been speculated that position-dependent quantum confinement effects can arise due to the hierarchical structures with highly faceted nanocrystals at the edges and tips of cubes, octahedrons and dendritic microstructures, resulting in several blue-shifted excitonic absorptions. This is also supported by the fact that the optical properties of non-spherical nanocrystals are controlled by the nanocrystals of lowest dimensions.30 Similar observations of blue-shift in the absorption maxima of PbS microstructures has been reported before.31,32
 |
| | Fig. 7 (a) Solid state absorbance spectra of uncapped PbS, S1–S5 and (b) MCE-capped PbS microstructures S6–S10. | |
The band gaps of the as-synthesized PbS microstructures were calculated from tauc plot and are given in ESI (Fig. S4†). The values of direct band gap energy (Eg) of PbS microstructures was estimated from a plot of (αhν)2 vs. photon energy (hν) following the Tauc relationship, αhν = A(hν − Eg)n where, hν = photon energy, A = constant, α = absorption coefficient, α = 4Πk/λ; k is the absorption index, λ is the wavelength and n depends on the type of transition, n = 1/2 for the allowed direct band gap. The first derivatives of the absorbance were plotted against the energy (eV). The inflection point (the point of the steepest curve) was recognised from the plot and the tangent in the Tauc plot at that inflection point is marked as the band gap of the sample.33 The value of band gap for S1–S10 fall in the range of 3.5–3.8 eV.
Photocatalytic properties
The photocatalytic degradation of toxic and non-biodegradable organic dyes using photocatalysts has attracted significant interest for the removal of environmental pollutants from waste water.34 In this regard, degradation of MO in aqueous solution with photocatalyst cubic (S2) and dendritic (S7) PbS microstructures was investigated under UV light irradiation. As MO dye is one of the major organic pollutants from textile and dyeing industries and it is carcinogenic and resistant to direct degradation by UV light or sunlight. However, it can be photoreduced to harmless organic compounds using photocatalysts such as PbS and the photodegradation of MO can be easily monitored with a decrease in the intensity of the characteristic absorption band at 463 nm. Therefore, the photocatalytic degradation of MO has gained significant interest. To rule out the possibility of decolouration by UV light, blank experiments were carried out in the absence of PbS photocatalyst which showed negligible degradation of MO suggesting the necessity of a photocatalyst for the degradation process. Fig. 8a and b show the changes in the optical absorption spectra of MO at different time intervals under UV light irradiation catalyzed by S2 (cubic) and S7 (dendritic) PbS respectively. As it can be seen from Fig. 8a, with an increase in irradiation time the concentration of MO decreases, and at the end of 180 min, the % degradation of MO was found to be 79.78 and 95.41% catalysed by cubic (S2) and dendritic (S7) PbS microstructures, respectively, indicating relatively higher photocatalytic performance of S7 (dendrites) over S2 (cubes) (Fig. 8c). Furthermore, the kinetics of photocatalytic degradation of MO using PbS microstructures can be best explained using a pseudo first order reaction, ln(Co/Ct) = kappt, where Co is the concentration of the dye after keeping it in the dark for 30 min and Ct is the concentration of the dye at given time interval t. The estimated values of the rate constant from the plot of ln(Co/Ct) vs. T (Fig. 8d) for the photocatalytic reaction catalysed by cubic (S2) and dendritic (S7) PbS samples were 1.13 × 10−2 and 2.45 × 10−2 min−1, respectively, suggesting higher photocatalytic activity of dendrites (S7) over cubes (S2). The higher activity of dendrites can be attributed to higher charge transfer possible due to its branched structure. Highly branched structures cause greater scattering of the light hence increases the free path for the light to travel.35 Greater is the scattering of light on the surface, higher will be the absorption because the scattered light will fall back on the branched structures results in increasing the concentration of the charge carries. Since charge carries govern the photocatalytic activity, the activity of branched structures will be higher as compared to the unbranched structures such as cubes.4a,36 Further, to determine the stability and recyclability of the photocatalysts, the PbS samples (S2 and S7) were isolated after the catalytic cycle by centrifuging the dispersed solution and were washed several times with methanol. The powder XRD spectra (Fig. S5, ESI†) of the recycled samples match very well with those of the original samples confirming the photostability and recyclability of both cubic (S2) and dendritic (S7) PbS samples. Moreover, the photocatalytic activity of S2 and S7 for degradation of MO is comparable to the activity reported for similar PbS microstructures in literature.37
 |
| | Fig. 8 (a) Time-dependent UV-vis absorption spectra for degradation of MO (2.0 × 10−5 M in 40 ml H2O) using PbS microstructures (25 mg S2) (a) and S7 (b) under UV light, (c) percentage conversion of MO with time (d) plot of ln(Co/C) with time. | |
Proposed mechanism of the degradation of MO
The mechanism of photocatalysis involves the absorption of a photon by the semiconducting material leading to excitation of electron from valence band (VB) to conduction band (CB) and formation of electron–hole (e−CB/h+VB) pair (as shown in eqn (1)).38| | |
PbS + hv → (e−CB)PbS + (h+VB)PbS
| (1) |
Eqn (2)–(5) describe the interaction of photogenerated electron–hole pair on the surface of the photocatalyst (PbS). It is crucial that the photogenerated electron–hole pair is transferred to the surface of the photocatalyst to inhibit the recombination of electrons and holes, thereby increasing the efficiency of the photocatalyst (PbS).
| | |
O2 + PbS(e−CB) → O2˙−
| (2) |
| | |
O2 + 2(e−CB)PbS + 2H+ → H2O2
| (3) |
| | |
(e−CB)PbS + O2˙− + 2H+ → HO˙ + HO−
| (4) |
| | |
H2O + (h+VB)PbS → HO˙ + H+
| (5) |
The reactive species (O2˙−, HO˙) formed react with MO dye to form the degradation product through several pathways as has been reported before (eqn (6) and (7).39–41
| | |
MO + radical species (O2˙−, HO˙)/H2O2 → degradationproducts
| (6) |
| | |
MO˙+ads → degradation products
| (7) |
Conclusions
In summary, a facile, one-pot, solvothermal route for the synthesis of template-free PbS microstructures composed of nanocrystals has been achieved with DBDS as a new temperature controlled in situ source of S2− ions. The interesting difference in the time-dependent morphological evolution of un-capped (S1–S5) and MCE-capped PbS microstructures has been reported. Further, the possible mechanism of the morphological evolution of PbS microstructures in the presence and absence of MCE capping agent has also been proposed. The morphology-dependent photocatalytic activity of cubic (S2) and dendritic (S7) PbS microstructures for the degradation of MO dye revealed higher photocatalytic activity of dendrites over cubes. The relatively higher activity of dendritic PbS microstructure as been attributed to better charge transfer originating due to its branched structure. Thus the influence of capping agent on the morphological evolution and the photocatalytic activity of PbS hierarchical structures has been presented.
Acknowledgements
C. M. N. gratefully acknowledges the financial support from Department of Science and Technology (DST), and Council of Scientific and Industrial Research (CSIR), Government of India. The authors are thankful to Mr Kausik Datta (ISM Dhanbad) for FESEM images.
References
-
(a) X. Liu, R. Huang and J. Zhu, Chem. Mater., 2008, 20, 192 CrossRef CAS;
(b) P. C. Ray, Chem. Rev., 2010, 110, 5332 CrossRef CAS PubMed;
(c) R. Yan, X. Sun, X. Wang, Q. Peng and Y. Li, Chem.–Eur. J., 2005, 11, 2183 CrossRef CAS PubMed.
-
(a) X. Wang, C. J. Summers and Z. L. Wang, Nano Lett., 2004, 4, 423 CrossRef CAS PubMed;
(b) S. Rabii, Phys. Rev., 1968, 167, 801 CrossRef CAS;
(c) L. A. Hemstreet, Phys. Rev., 1975, 11, 2260 CrossRef CAS;
(d) W. W. Scanlon, J. Phys. Chem. Solids, 1959, 8, 423 CrossRef CAS;
(e) Y. Hu, Z. Jiang, C. Xu, T. Mei, J. Guo and T. White, J. Phys. Chem. C, 2007, 111, 9757 CrossRef CAS.
-
(a) L. Zhao, H. Zhang, Y. Xing, S. Song, S. Yu, W. Shi, X. Guo, J. Yang, Y. Lei and F. Cao, Chem. Mater., 2008, 20, 198 CrossRef CAS;
(b) Q. Song and Z. J. Zhang, J. Am. Chem. Soc., 2004, 126, 6164 CrossRef CAS PubMed;
(c) G. Y. Yurkova, A. S. Fionovb, Y. A. Koksharovc, V. V. Kolesovb and S. P. Gubina, Inorg. Mater., 2007, 43, 834 CrossRef.
-
(a) R. Boppella, K. Anjaneyulu, P. Basak and S. V. Manorama, J. Phys. Chem. C, 2013, 117, 4597 CrossRef CAS;
(b) Z. Y. Zhou, N. Tian, J. T. Li, I. Broadwell and S. G. Sun, Chem. Soc. Rev., 2011, 40, 4167 RSC.
-
(a) L. Bakueva, I. Gorelikov, S. Musikhin, X. S. Zhao, E. H. Sargent and E. Kumacheva, Adv. Mater., 2004, 16, 926 CrossRef CAS;
(b) K. R. Choudhury, Y. Sahoo, S. Jang and P. N. Prasad, Adv. Funct. Mater., 2005, 15, 751 CrossRef;
(c) H. S. Kim, M. H. Lee, N. C. Jeong, S. M. Lee, B. K. Rhee and K. B. Yoon, J. Am. Chem. Soc., 2006, 128, 15070 CrossRef CAS PubMed.
-
(a) C. Zhang, Z. Kang, E. Shen, E. Wang, L. Gao, F. Luo, C. Tian, C. Wang and Y. Lan, J. Phys. Chem. B, 2006, 110, 184 CrossRef CAS PubMed;
(b) Y. F. Li, M. Zhang, Q. J. Yang, F. X. Zhang, M. Q. Zheng and A. J. Wang, J. Nanosci., 2015, 2015, 362023 Search PubMed , pp 9.
-
(a) J. P. Ge, J. Wang, H. X. Zhang, X. Wang, Q. Peng and Y. D. Li, Chem.–Eur. J., 2005, 11, 1889 CrossRef CAS PubMed;
(b) P. L. Nichols, M. Sun and C. Z. Ning, ACS Nano, 2011, 5, 8730 CrossRef CAS PubMed.
- P. T. Zhao, G. Chen, Y. Hu, X. L. He, K. Wu, Y. Cheng and K. X. Huang, J. Cryst. Growth, 2007, 303, 632 CrossRef CAS.
- H. S. Chen, S. C. Wu and M. H. Huang, Dalton Trans., 2015, 44, 15088 RSC.
- Y. Wang, Q. Dai, X. Yang, B. Zou, D. Li, B. Liu, M. Z. Hub and G. Zoua, CrystEngComm, 2011, 13, 199 RSC.
- S. M. Lee, Y. W. Jun, S. N. Cho and J. Cheon, J. Am. Chem. Soc., 2002, 124, 11244 CrossRef CAS PubMed.
- M. J. Bierman, Y. K. A. Lau and S. Jin, Nano Lett., 2007, 7, 2907 CrossRef CAS PubMed.
- B. Deng, S. L. Zhong, D. H. Wang, S. S. Wang, T. K. Zhang, W. G. Qua and A. W. Xu, Nanoscale, 2011, 3, 1014 RSC.
- D. Kuang, A. Xu, Y. Fang, H. Liu, C. Frommen and D. Fenske, Adv. Mater., 2003, 15, 1747 CrossRef CAS; S. Xiong, B. Xi, D. Xu, C. Wang, X. Feng, H. Zhou and Y. Qian, J. Phys. Chem. C, 2007, 111, 16761 Search PubMed.
-
(a) G. J. Zhou, M. K. Lu, Z. L. Xiu, S. F. Wang, H. P. Zhang, Y. Y. Zhou and S. M. Wang, J. Phys. Chem. B, 2006, 110, 6543 CrossRef CAS PubMed;
(b) Y. Jiang, Y. Wu, B. Xie, S. W. Yuan, X. M. Liu and Y. T. Qian, J. Cryst. Growth, 2001, 231, 248 CrossRef CAS;
(c) B. Ding, M. Shi, F. Chen, R. Zhou, M. Deng, M. Wang and H. Chen, J. Cryst. Growth, 2009, 311, 1533 CrossRef CAS;
(d) A. A. Patel, F. Wu, J. Z. Zhang, C. L. Torres-Martinez, R. K. Mehra, Y. Yang and S. H. Risbud, J. Phys. Chem. B, 2000, 104, 11598 CrossRef CAS;
(e) Y. Wang, X. Yang, G. Xiao, B. Zhou, B. Liu, G. Zou and B. Zou, CrystEngComm, 2013, 15, 5496 RSC;
(f) H. Li, D. Chen, L. Li, F. Tang, L. Zhanga and J. Rena, CrystEngComm, 2010, 12, 1127 RSC;
(g) Y. Hou, H. Kondoh and T. Ohta, Cryst. Growth Des., 2009, 9, 3119 CrossRef CAS.
-
(a) R. Jin, G. Chen, Q. Wang and J. Pei, Mater. Lett., 2011, 65, 1151 CrossRef CAS;
(b) F. Li, X. Huanga, T. Kong, X. Liub, Q. Qinb and Z. Li, J. Alloys Compd., 2009, 485, 554 CrossRef CAS;
(c) Q. L. Huang, H. Chen, C. L. Wu and Y. C. Zhang, Mater. Lett., 2010, 64, 1891 CrossRef CAS;
(d) W. Qiu, M. Xu, F. Chen, X. Yang, Y. Nana and H. Chen, CrystEngComm, 2011, 13, 4689 RSC;
(e) F. Chen, W. Qiu, X. Chen, M. Wanga and H. Chen, CrystEngComm, 2010, 12, 1893 RSC.
-
(a) C. M. Nagaraja and M. Kaur, Mater. Lett., 2013, 111, 230 CrossRef CAS;
(b) M. Kaur and C. M. Nagaraja, RSC Adv., 2014, 4, 18257 RSC;
(c) M. Kaur, N. K. Gupta and C. M. Nagaraja, CrystEngComm, 2015, 17, 2359 RSC;
(d) M. Kaur and C. M. Nagaraja, Mater. Lett., 2015, 154, 90 CrossRef CAS.
- T. Mandal, G. Piburn, V. Stavila, I. Rusakova, T. Ould-Ely, A. C. Colson and K. H. Whitmire, Chem. Mater., 2011, 23, 4158 CrossRef CAS.
- Y. W. Jun, J. H. Lee, J. S. Choi and J. Cheon, J. Phys. Chem. B, 2005, 109, 14795 CrossRef CAS PubMed.
-
(a) Z. L. Wang, J. Phys. Chem. B, 2000, 104, 1153 CrossRef CAS;
(b) Y. Cao, P. Hu and D. Jia, Nanoscale Res. Lett., 2012, 7, 668 CrossRef PubMed.
- M. Liu, M. Leng, D. Liu, F. Chen, C. Li and C. Wang, Inorg. Chem., 2014, 53, 11484 CrossRef CAS PubMed.
- I. Sunagawa, Crystals: Growth, Morphology, and Perfection, Cambridge University Press, Cambridge, U.K, 2005 Search PubMed.
-
(a) W. F. Berg, Crystal growth from solution, Proc. R. Soc. London, Ser. A, 1938, 164, 79–95 CrossRef CAS;
(b) M. Jin, H. Zhang, Z. Xie and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 7850 CrossRef CAS PubMed.
-
(a) T. J. Zhu, X. Chen, Y. Q. Cao and X. B. Zhao, J. Phys. Chem. C, 2009, 113, 8085 CrossRef CAS;
(b) A. Q. Fernandez, J. C. H. Garrido, H. Yang, Y. Zhou, A. Varela, M. Parras, J. J. C. Gamez, J. M. G. Calbet, P. F. Green and N. A. Kotov, ACS Nano, 2012, 6, 3800 CrossRef PubMed.
- A. J. Wang, Q. C. Liao, J. J. Feng, P. P. Zhang, Z. M. Zhang and J. R. Chen, Cryst. Growth Des., 2012, 12, 832 CAS.
- Z. Quan, C. Li, X. Zhang, J. Yang, P. Yang, C. Zhang and J. Lin, Cryst. Growth Des., 2008, 8, 2384 CAS.
- P. Zhao, J. Wang, G. Cheng and K. Huang, J. Phys. Chem. B, 2006, 110, 22400 CrossRef CAS PubMed.
- H. Weller, Angew. Chem., Int. Ed., 1993, 32, 41 CrossRef.
- Y. Ma, L. Qi, J. Ma and H. Cheng, Cryst. Growth Des., 2004, 4, 351 CAS.
-
(a) K. K. Nanda and S. N. Dahu, Adv. Mater., 2001, 13, 280 CrossRef CAS;
(b) C. Li, Y. Zhao, F. Li, Z. Shi and S. Feng, Chem. Mater., 2010, 22, 1901 CrossRef CAS.
- N. Zhou and L. Qui, Adv. Mater., 2006, 18, 359 CrossRef.
- W. S. Chae, H. W. Shin, E. S. Lee, E. J. Shin, J. S. Jung and Y. R. Kim, J. Phys. Chem. B, 2005, 109, 6204 CrossRef CAS PubMed.
- A. E. Morales, E. S. Mora and U. Pal, Rev. Mex. Fis. S, 2007, 53, 18 CAS.
- S. Kundu, M. D. Mukadam, S. M. Yusuf and M. Jayachandran, CrystEngComm, 2013, 15, 482 RSC.
- M. J. Bierman and S. Jin, Energy Environ. Sci., 2009, 2, 1050 CAS.
-
(a) P. Dong, Y. Wang, H. Li, H. Li, X. Ma and L. Han, J. Mater. Chem. A., 2013, 1, 4651 RSC;
(b) F. X. Bu, M. Hu, L. Xu, Q. Meng, G. Y. Mao, D. M. Jiang and J. S. Jiang, Chem. Commun., 2014, 50, 8543 RSC.
-
(a) A. Pourahmad, Synth. React. Inorg. Met.-Org. Chem., 2015, 45, 1080 CrossRef CAS;
(b) A. V. Borhadea and B. K. Uphade, Chalcogenide Lett., 2012, 9, 299 Search PubMed;
(c) I. Yadav, S. Nihalaniand and S. Bhardwaj, Chem. Sin., 2012, 3, 1468 Search PubMed.
- H. Zhang and Y. Zhu, J. Phys. Chem. C, 2010, 114, 5822 CAS.
- P. Raja, A. Bozzi, H. Mansilla and J. Kiwi, J. Photochem. Photobiol., A, 2005, 169, 271 CrossRef CAS.
- M. Stylidi, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2004, 47, 189 CrossRef CAS.
- A. H. Boonstra and C. A. H. A. Mutsaers, J. Phys. Chem., 1975, 79, 1940 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra09110e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.