
Base-mediated regiospecific cascade synthesis of N-(2-pyridyl)pyrroles from N-propargylic β-enaminones†
Jinhai Shena,
Xifa Yanga,
Fuyuan Wanga,
Yue Wangb,
Guolin Chenga and
Xiuling Cui*a
aKey Laboratory of Xiamen Marine and Gene Drugs, Institutes of Molecular Medicine and School of Biomedical Sciences, Huaqiao University & Engineering Research Center of Molecular Medicine, Ministry of Education, Xiamen 361021, China. E-mail: cuixl@hqu.edu.cn
bDepartment of Chemistry, SUNY Stony Brook, Stony Brook, NY 11794-3400, USA
First published on 12th May 2016
Abstract
A KOH-promoted regiospecific synthesis of polysubstituted N-(2-pyridyl)pyrroles under transition metal-free conditions has been developed. The pyrrole and pyridine rings were simultaneously installed from acyclic enaminone precursors under mild conditions and generated 1 equiv. of H2O as the sole byproduct. A series of polysubstituted N-(2-pyridyl)pyrroles were provided in up to 91% yield for 21 examples.
N-(2-Pyridyl)pyrrole is an important heterocycle and key skeleton in natural products, pharmaceuticals and advanced functional materials,1 which are exemplified by the four examples shown in Fig. 1. Consequently, developing mild and efficient access to N-(2-pyridyl)pyrroles is of great significance.
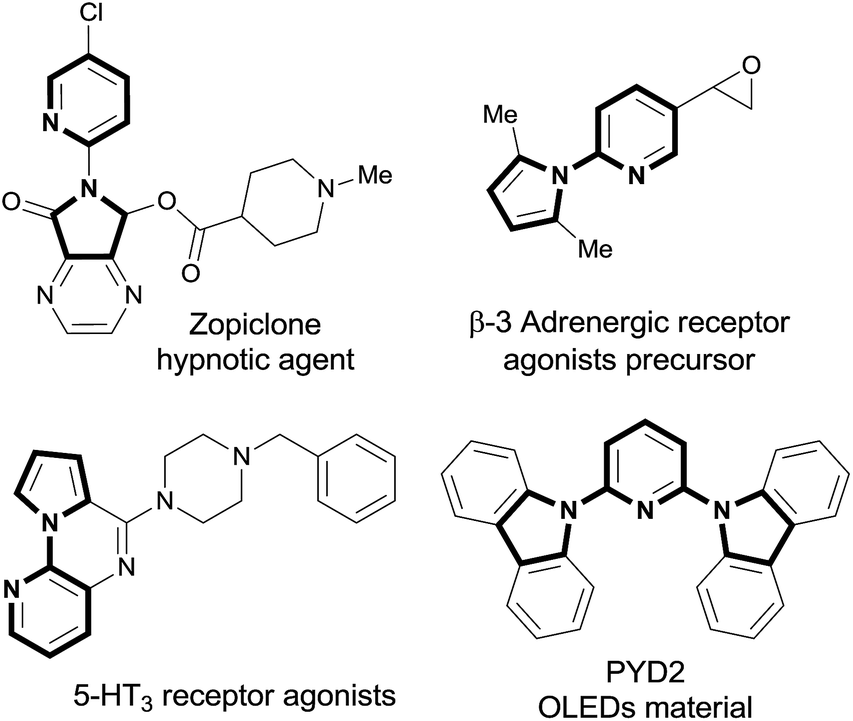 | ||
| Fig. 1 Four examples illustrating the importance of the N-(2-pyridyl)pyrrole. | ||
Conventionally, N-(2-pyridyl)pyrroles have been synthesized by the Paal–Knorr reaction from 2-amino-pyridines with 1,4-dicarbonyl compounds2 (or 2,5-dimethoxytetrahydrofuran3). Recently, copper-catalysed Ullmann-type coupling of 2-halopyridines with pyrroles has been developed as a straightforward method to construct such a structure.4 However, in this procedure the pyridine scaffold and the pyrrole ring need to be constructed in advance. Moreover, the requirement of a transition metal may cause contamination of the products that limits their applications, especially in the pharmaceutical industry.5 Therefore, a general and metal-free procedure to synthesize N-(2-pyridyl)pyrroles under mild reaction conditions remains highly desirable.
Recently, cascade reactions have received much attention for the assembly of complicated molecules from relatively simple starting materials in an economically favourable process.6 Among which, the interception of in situ generated reactive species has been perceived as a powerful shortcut for the discovery of new cascade reactions with high efficiency. Due to the presence of nucleo/electro-philic enaminone7 and a highly reactive C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bond, N-propargylic β-enaminones 1 may be a potential precursor of cascade reactions and their utilization in organic synthesis remains a subject of great current interest.8 In 2008, Cacchi and co-workers reported that N-propargylic β-enaminones 1 could be selectively transformed into pyrroles via a base-promoted 5-exo-dig cyclization (Scheme 1, 3).9 According to our previous work,10 1,4-oxazepines can also be formed via a 7-exo-dig cyclization under basic conditions (Scheme 1, 4). We assumed that the highly reactive species 4 might be trapped by the pyrrole 3 (formed in situ) to give N-(2-pyrcidyl)pyrroles 2 in the presence of a suitable base. In continuation of our effort on enaminone chemistry,11 herein we describe a base-mediated regiospecific synthesis of N-(2-pyridyl)pyrroles from N-propargylic enaminones (Scheme 1). As far as we know, this is the first report to simultaneously build pyrrole and pyridine rings from an acyclic precursor under metal-free conditions.12
C triple bond, N-propargylic β-enaminones 1 may be a potential precursor of cascade reactions and their utilization in organic synthesis remains a subject of great current interest.8 In 2008, Cacchi and co-workers reported that N-propargylic β-enaminones 1 could be selectively transformed into pyrroles via a base-promoted 5-exo-dig cyclization (Scheme 1, 3).9 According to our previous work,10 1,4-oxazepines can also be formed via a 7-exo-dig cyclization under basic conditions (Scheme 1, 4). We assumed that the highly reactive species 4 might be trapped by the pyrrole 3 (formed in situ) to give N-(2-pyrcidyl)pyrroles 2 in the presence of a suitable base. In continuation of our effort on enaminone chemistry,11 herein we describe a base-mediated regiospecific synthesis of N-(2-pyridyl)pyrroles from N-propargylic enaminones (Scheme 1). As far as we know, this is the first report to simultaneously build pyrrole and pyridine rings from an acyclic precursor under metal-free conditions.12
 | ||
| Scheme 1 Proposed route to N-(2-pyridyl)pyrroles. | ||
1,3-Diphenyl-3-(prop-2-yn-1-ylamino)-2-propen-1-one 1a was chosen as a model substrate to optimize the reaction parameters. Initially, the model reaction of 1a was examined using KOH/DMSO system, which was used as a base/solvent system for N-pyridylation of heteroarenes in our previous report.10 To our delight, the desired N-(2-pyridyl)pyrrole product 2a was obtained in 28% yield as well as pyrrole 3a in 40% yield. The structure of 2a was confirmed by single crystal X-ray diffraction (see ESI† for details). Then a series of solvents was screened, and the results showed that aprotic solvents favoured the formation of the desired product 2a and CH3CN turned out to be the most effective solvent (Table 1, entries 1–7). The KOH/DMSO system, being of stronger basicity,13 may favour the formation of pyrrole 3a through the 5-exo-dig cyclization, which resulted in a lower yield of 2a. Further investigation on the influence of base supported our hypothesis. Acceptable yields of 2a were achieved in the presence of Cs2CO3 and NaOH (Table 1, entries 8 and 9). High-alkaline bases, such as NaOtBu and KOtBu, gave pyrrole 3a as main product (35% and 43%, respectively, entries 10 and 11). In contrast, no reactions occurred when weaker bases, such as K2CO3, Et3N or DABCO (1,4-diaza[2.2.2]bicyclooctane), were used (Table 1, entries 12–14). Decreasing the amount of KOH from 2 equiv. to 1.5 equiv. resulted in a lower yield of 2a (Table 1, entry 15). Remarkably, a respectable 62% yield of 2a was achieved using 0.3 equiv. of KOH (entry 16). The requirement of high loading of base might be due to the low solubility of KOH in CH3CN.14 Decreasing reaction temperature resulted in a lower yield (Table 1, entry 17). When a radical-trapping reagent, 2,2,6,6-tetramethylpiperidinooxy (TEMPO), was added to the reaction, the yield of the product was not influenced significantly, indicating that the radical pathway is not likely to be involved in this transformation (Table 1, entry 18). Under N2 atmosphere, the reaction provided a similar result (entry 19 vs. 7). The starting substrate remains unreactive without base (Table 1, entry 20). To avoid the involvement of transition metals in the reaction, KOH (99.999% purity) was used instead of a purity of 98%, providing a similar yield (92%, entry 21 vs. 7), indicating that the purity of KOH was irrelevant here. Importantly, the base could be simply recovered and reused by simple filtration after the reactions was finished. The reactivity of KOH gradually lost during the eight recycles (Fig. 2).
|
||||
|---|---|---|---|---|
| Entry | Base | Solvent | Temp (°C) | Yieldb (%) 2a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3a:4a :3a:4a |
| a Reaction conditions: 1a (0.5 mmol), and base (1 mmol) in solvent (2 mL) at corresponding temperature for 30 min.b Isolated yield.c 0.75 mmol of base was used.d 0.15 mmol of base was used for 2 h.e 2.0 equiv. of TEMPO (2,2,6,6-tetramethylpiperidinooxy) was added.f Under N2 atmosphere.g Use highly pure KOH (99.999% purity). DMF = N,N-dimethylformamide. DMSO = dimethylsulfoxide. | ||||
| 1 | KOH | DMSO | 120 | 28:40:0 |
| 2 | KOH | DMF | 120 | 85:0:0 |
| 3 | KOH | DMAc | 120 | 86:0:0 |
| 4 | KOH | Dioxane | Reflux | Trace:45:0 |
| 5 | KOH | EtOH | Reflux | 0:12:0 |
| 6 | KOH | Toluene | Reflux | 0:0:0 |
| 7 | KOH | CH3CN | Reflux | 91:0:trace |
| 8 | Cs2CO3 | CH3CN | Reflux | 85:trace:0 |
| 9 | NaOH | CH3CN | Reflux | 79:8:0 |
| 10 | NaOtBu | CH3CN | Reflux | 30:35:0 |
| 11 | KOtBu | CH3CN | Reflux | 23:43:0 |
| 12 | K2CO3 | CH3CN | Reflux | 0:0:0 |
| 13 | Et3N | CH3CN | Reflux | 0:0:0 |
| 14 | DABCO | CH3CN | Reflux | 0:0:0 |
| 15c | KOH | CH3CN | Reflux | 82:0:10 |
| 16d | KOH | CH3CN | Reflux | 62:0:16 |
| 17 | KOH | CH3CN | 60 | 78:0:11 |
| 18e | KOH | CH3CN | Reflux | 90:0:trace |
| 19f | KOH | CH3CN | Reflux | 90:0:trace |
| 20 | — | CH3CN | Reflux | 0:0:0 |
| 21g | KOH | CH3CN | Reflux | 92:0:trace |

 | ||
| Fig. 2 Recycling KOH. | ||
The scope and generality of the substrates for this process was next investigated under the optimized reaction conditions (Table 1, entry 7). As shown in Table 2, R1 in substrate 1 could be either electron-rich or electron-deficient aryl groups, and provided the corresponding N-(2-pyridyl)pyrroles in 64–89% yields (entries 1–10). Due to steric hindrance, the substrates with para-substituents gave slightly higher yields than those with ortho-substituents (entries 3 and 5 vs. entries 2 and 4). Halogens, such as F, Cl and Br were all tolerated well, which made this reaction particularly attractive for increasing the molecular complexity by transition-metal-catalyzed coupling reactions. Moreover, when R1 is a heteroaryl group, such as 2-thienyl, this transformation still could proceed smoothly and gave the multi-heterocycle product 2k in 69% yield (entry 11). Alkyl groups, such as isopropyl- and cyclohexyl-, were also tolerated, although giving the desired products in declined yields (entries 12 and 13).
|
|||||
|---|---|---|---|---|---|
| Entry | 1 | R1 | R2 | 2 | Yieldb (%) |
| a Reactions were carried out in open air on a 0.5 mmol scale using 2 equiv. of KOH in 2 mL of CH3CN under refluxing for 30 min.b Isolated yield. | |||||
| 1 | 1a | Ph | Ph | 2a | 91 |
| 2 | 1b | 2-MeC6H4- | Ph | 2b | 78 |
| 3 | 1c | 4-MeC6H4- | Ph | 2c | 81 |
| 4 | 1d | 2-BrC6H4- | Ph | 2d | 75 |
| 5 | 1e | 4-BrC6H4- | Ph | 2e | 81 |
| 6 | 1f | 4-ClC6H4- | Ph | 2f | 77 |
| 7 | 1g | 4-FC6H4- | Ph | 2g | 76 |
| 8 | 1h | 4-MeOC6H44- | Ph | 2h | 80 |
| 9 | 1i | 4-CF3C6H4- | Ph | 2i | 70 |
| 10 | 1j | Naphthalen-2- | Ph | 2j | 85 |
| 11 | 1k | Thiophen-2- | Ph | 2k | 69 |
| 12 | 1l | Isopropyl- | Ph | 2l | 28 |
| 13 | 1m | Cyclohexyl- | Ph | 2m | 45 |
| 14 | 1n | Ph | 2-MeC6H4- | 2n | 73 |
| 15 | 1o | Ph | 3-MeC6H4- | 2o | 85 |
| 16 | 1p | Ph | 4-MeC6H4- | 2p | 91 |
| 17 | 1q | Ph | 4-BrC6H4- | 2q | 74 |
| 18 | 1r | Ph | 4-ClC6H4- | 2r | 71 |
| 19 | 1s | Ph | 4-FC6H4- | 2s | 88 |
| 20 | 1t | Ph | 4-MeOC6H4- | 2t | 78 |
| 21 | 1u | Ph | Thiophen-2- | 2u | 75 |


The scope of the R2 group in the substrate was also investigated. In general, R2 could be electron-rich or electron-deficient aryls or heteroaryls (entries 14–21). The steric hindrance did not significantly affect this reaction. Substrates with ortho-, meta-, or para-methyl substituents did not diminish the efficiency of this transformation (entries 14–16). Heterocyclic substituents are also suitable, albeit with slightly lower efficacy (entry 21). When R2 is alkyl group, such as n-butyl, the substrate was decomposed. No main product could be obtained. Free acetylene group is essential to get the desired product. When phenyl and methyl-tethered N-propargylic β-enaminones, 3-((3-(4-methoxyphenyl)prop-2-yn-1-yl)amino)-1,3-diphenylprop-2-en-1-one 5 and 3-(but-2-yn-1-ylamino)-1,3-diphenylprop-2-en-1-one 7, were employed as starting materials in this reaction, only pyrrole 6 and 8 were afforded via base-promoted 5-exo-dig cyclization (eqn (1) and (2)).
In order to gain insight into the mechanism, some controlled experiments were carried out (Scheme 2). When the reaction of N-propargylic β-enaminone 1a under standard conditions was quenched by water after 2 min, besides the desired product 2a, pyrrole 3a and 1,4-oxazepine 4a were obtained in 21% and 13% yields, respectively (eqn (3)). The reaction of pyrrole 3a and 1,4-oxazepine 4a under the standard conditions gave the desired product 2a in 94% yield (eqn (4)), suggesting pyrroles 3 and 1,4-oxazepines 4 were generated in situ and involved in this cascade reaction. Moreover, when an external nucleophile 9 was added into the reaction of 1a under the standard conditions, N-(2-pyridyl)pyrrole product 2a was obtained in 18% yield, as well as N-(2-pyridyl)indole 10 in 34% yield and pyrrole 3a in 28% yield (eqn (5)), indicating that pyrrole 3a and 1,4-oxazepine 4a were formed in roughly equal amounts during the process of the reaction.
 | ||
| Scheme 2 Controlled experiment. | ||
On the basis of the aforementioned observations and our previous work,10 a tentative mechanism for the formation of functionalized N-(2-pyridyl)pyrroles 2 was proposed, as depicted in Scheme 3. On one hand, base-promoted 5-exo-dig cyclization of 1 provided the substituted pyrrole scaffolds 3. On the other hand, base-mediated propargyl–allenyl isomerisation and enolization of 1 generated iminoenolate intermediates A, followed by intramolecular 7-exo-dig cyclization to afford 1,4-oxazepines 4, which subsequently underwent a 6π-electrocyclization and a walk rearrangement to give epoxide intermediates C. SN2 attack of pyrrole anions D resulted in an epoxide ring-opening of C to generate trans-2,3-dihydropyridine intermediates E. Finally, the protonation and dehydrative aromatization led to the desired products 2.
 | ||
| Scheme 3 Plausible reaction mechanism. | ||
Conclusions
In summary, we have described a novel and highly efficient base-promoted cascade reaction for the synthesis of the diverse N-(2-pyridyl)pyrroles from N-propargylic β-enaminones by tuning the basicity of reaction conditions. This procedure simultaneously facilitated the construction of a pyridine scaffold and generation of a pyrrole ring, and tolerated a wide range of functional groups. The formation of 1,4-oxazepines, and interception by the pyrroles generated in situ were key steps in this metal-free cascade reaction. This protocol only required 2 equiv. of KOH as an additive, and generated 1 equiv. of H2O as the sole byproduct, thereby making this process atom economic and environmentally friendly.Acknowledgements
This research was supported by NSF of China (21572072), Xiamen Southern Oceanographic Center (15PYY052SF01) and Huaqiao University.Notes and references
- (a) W. J. Hudak, C. H. Tilford and R. E. Lewis, J. Med. Chem., 1971, 14, 328 CrossRef CAS PubMed; (b) M. E. H. Amrani, M. S. Ibrahim and K. C. Persaud, Mater. Sci. Eng., C, 1993, 1, 17 CrossRef; (c) Y. Mo, F. Bai and Z. Wang, J. Photochem. Photobiol., A, 1995, 92, 25 CrossRef CAS; (d) B. Kovač, L. Klasinc, J. Vorkapić-Furač, M. Mintas and J. Knop, J. Chem. Soc., Perkin Trans. 2, 1997, 12, 2597 RSC; (e) S. P. Sibley, A. Saunders, P. Crum, K. Mutolo, J. L. Menke and E. V. Patterson, J. Photochem. Photobiol., A, 2004, 163, 463 CrossRef CAS; (f) R. W. Scott, D. E. Fox, J. W. Wong and M. P. Burns, Org. Process Res. Dev., 2004, 8, 583 CrossRef; (g) P. Ray, J. Wright, J. Adam, J. Bennett, S. Boucharens, D. Black, A. Cook, A. R. Brown, O. Epemolu and D. Fletcher, Bioorg. Med. Chem. Lett., 2011, 21, 7 Search PubMed; (h) X.-H. Zhao, Z.-S. Zhang, Y. Qian, M.-D. Yi, L.-H. Xie, C.-P. Hu, G.-H. Xie, H. Xu, C.-M. Han and Y. Zhao, J. Mater. Chem. C, 2013, 1, 3482 RSC.
- (a) L. Knorr, Chem. Ber., 1884, 17, 1635 CrossRef; (b) S. K. De, Heteroat. Chem., 2008, 19, 592 CrossRef CAS.
- (a) D. Bandyopadhyay, S. Mukherjee, J. C. Granados, J. D. Short and B. K. Banik, Eur. J. Med. Chem., 2012, 50, 209 CrossRef CAS PubMed; (b) M. A. Wilson, G. Filzen and G. S. Welmaker, Tetrahedron Lett., 2009, 50, 4807 CrossRef CAS.
- (a) F. Ullmann, Ber. Dtsch. Chem. Ges., 1903, 36, 2382 CrossRef; (b) J. Lindley, Tetrahedron, 1984, 40, 1433 CrossRef CAS; (c) J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed; (d) M. Taillefer, N. Xia and A. Ouali, Angew. Chem., Int. Ed., 2007, 46, 934 CrossRef CAS PubMed; (e) M. Schnurch, R. Flasik, A. F. Khan, M. Spina, M. D. Mihovilovic and P. Stanetty, Eur. J. Org. Chem., 2006, 3283 CrossRef CAS; (f) H.-C. Ma and X.-Z. Jiang, J. Org. Chem., 2007, 72, 8943 CrossRef CAS PubMed; (g) Y.-C. Teo, F.-F. Yong and S. Sim, Tetrahedron, 2013, 69, 7279 CrossRef CAS.
- (a) F.-X. Felpin, T. Ayad and S. Mitra, Eur. J. Org. Chem., 2006, 2679 CrossRef CAS; (b) J. Mao, Q. Hua, G. Xie, J. Guo, Z. Yao, D. Shi and S. Jia, Adv. Synth. Catal., 2009, 351, 635 CrossRef CAS; (c) C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044 CrossRef CAS PubMed; (d) W. Liu, H. Cao, H. Zhang, H. Zhang, K. H. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737 CrossRef CAS PubMed.
- For recent reviews, see: (a) J. C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001 CrossRef CAS PubMed; (b) K. C. Nicolaou and J. S. Chen, Chem. Soc. Rev., 2009, 38, 2993 RSC; (c) H. Pellissier, Chem. Rev., 2013, 113, 442 CrossRef CAS PubMed; (d) C. M. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed.
- For recent reviews, see: (a) A.-Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463 CrossRef CAS; (b) B. Stanovnik and J. Svete, Chem. Rev., 2004, 104, 2433 CrossRef CAS PubMed; (c) B. Govindh, B. S. Diwakar and Y. L. Murthy, Org. Commun., 2012, 5, 105 Search PubMed; (d) A. K. Chattopadhyay and S. Hanessian, Chem. Commun., 2015, 51, 16437 RSC; (e) A. K. Chattopadhyay and S. Hanessian, Chem. Commun., 2015, 51, 16450 RSC.
- For some recent references, see: (a) A. Saito, T. Konishi and Y. Hanzawa, Org. Lett., 2009, 12, 372 CrossRef PubMed; (b) C. Jiang, M. Xu, S. Wang, H. Wang and Z. J. Yao, J. Org. Chem., 2010, 75, 4323 CrossRef CAS PubMed; (c) K. K. Toh, Y.-F. Wang, E. P. Jian Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 13942 CrossRef CAS PubMed; (d) X. Xin, D. Wang, F. Wu, X. Li and B. Wan, J. Org. Chem., 2013, 78, 4065 CrossRef CAS PubMed; (e) M. A. Martins, M. Rossatto, C. P. Frizzo, E. Scapin, L. Buriol, N. Zanatta and H. G. Bonacorso, Tetrahedron Lett., 2013, 54, 847 CrossRef CAS; (f) K. Goutham, N. S. V. M. Rao Mangina, S. Suresh, P. Raghavaiah and G. V. Karunakar, Org. Biomol. Chem., 2014, 12, 2869 RSC; (g) K. Goutham, V. Nagaraju, S. i Suresh, P. Raghavaiah and G. V. Karunakar, RSC Adv., 2014, 4, 21054 RSC; (h) S. Karabiyikoglu, Y. Kelgokmen and M. Zora, Tetrahedron, 2015, 71, 4324 CrossRef CAS; (i) K. Goutham, D. Ashok Kumar, S. Suresh, B. Sridhar, R. Narender and G. V. Karunakar, J. Org. Chem., 2015, 80, 11162 CrossRef CAS PubMed.
- S. Cacchi, G. Fabrizi and E. Filisti, Org. Lett., 2008, 10, 2629 CrossRef CAS PubMed.
- G. Cheng, Y. Weng, X. Yang and X. Cui, Org. Lett., 2015, 17, 3790 CrossRef CAS PubMed.
- (a) G. Cheng, X. Zeng, J. Shen, X. Wang and X. Cui, Angew. Chem., Int. Ed., 2013, 52, 13265 CrossRef CAS PubMed; (b) J. Shen, G. Cheng and X. Cui, Chem. Commun., 2013, 49, 10641 RSC; (c) J. Shen, C. Ding, C. Kuai, Y. Liu, M. Wei, G. Cheng and X. Cui, J. Org. Chem., 2015, 80, 6584 CrossRef CAS PubMed; (d) J. Shen, L. Xue, X. Lin, G. Cheng and X. Cui, Chem. Commun., 2016, 52, 3293 Search PubMed; (e) J. Shen, X. Wang, X. Lin, Z. Yang, G. Cheng and X. Cui, Org. Lett., 2016, 18, 1378 CrossRef CAS PubMed.
- For reviews, see: (a) M. Malacria, Chem. Rev., 1996, 96, 289 CrossRef CAS PubMed; (b) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199 CrossRef CAS PubMed; (c) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS PubMed; (d) W. A. van Otterlo and C. B. De Koning, Chem. Rev., 2009, 109, 3743 CrossRef CAS PubMed; (e) A. V. Gulevich, A. S. Dudnik, N. Chernyak and V. Gevorgyan, Chem. Rev., 2013, 113, 3084 CrossRef CAS PubMed; (f) R. A. Foster and M. C. Willis, Chem. Soc. Rev., 2013, 42, 63 RSC.
- (a) B. A. Trofimov, Sulfur Rep., 1992, 74, 207 CrossRef; (b) R. Cano, D. J. Ramon and M. Yus, J. Org. Chem., 2011, 76, 654 CrossRef CAS PubMed; (c) Y. Fang, Y. Zheng and Z. Wang, Eur. J. Org. Chem., 2012, 77, 1495 CrossRef; (d) M. Joshi, M. Patel, R. Tiwari and A. K. Verma, J. Org. Chem., 2012, 77, 5633 CrossRef CAS PubMed.
- The solubility of KOH in MeCN under reflux was measured to be 0.02 (g/100 g), for examples showing the low solubility of KOH in MeCN, see: (a) S. A. DiBiase, J. R. Beadle and G. W. Gokel, Org. Synth., 1984, 62, 179 CrossRef CAS; (b) M. Zhao, W. Zhang and Z. Shen, J. Org. Chem., 2015, 80, 8868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 994843 and 994844. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra08987a |
| This journal is © The Royal Society of Chemistry 2016 |