
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A DFT study of the adsorption of O2 and H2O on Al(111) surfaces
Xin
Wei
a,
Chaofang
Dong
*a,
Zhanghua
Chen
b,
Kui
Xiao
a and
Xiaogang
Li
a
aCorrosion and Protection Center, Key Laboratory for Corrosion and Protection (MOE), University of Science and Technology Beijing, Beijing 100083, China. E-mail: cfdong@ustb.edu.cn; Fax: +86-10-62334005; Tel: +86-10-62333931 ext. 518
bSchool of Mathematics and Physics, University of Science and Technology Beijing, Beijing, 100083, China
First published on 6th June 2016
Abstract
Using first-principles calculations that are based on density functional theory, the molecular and dissociative adsorptions of O2 and H2O on a clean and O pre-adsorbed Al(111) surface were systematically investigated. The van der Waals dispersion correction is considered for the molecular adsorption of H2O. We found that O2 dissociates into O atoms which can adsorb on fcc and hcp sites. The stability ranking for O atoms on the clean Al(111) surface is fcc > hcp. The energy barriers for the migration of a single O atom from a hcp to a fcc site on a clean and O pre-adsorbed Al(111) surface are 25.91 kJ mol−1 and 28.67 kJ mol−1, respectively, which means that the pre-adsorbed O atom inhibits the migration of O atoms on the surface. H2O molecules cannot dissociate on both clean and O pre-adsorbed Al(111) surfaces spontaneously. The pre-adsorbed O atom can strengthen the adsorption of H2O and promote its deformation. The dissociation adsorption of H2O, that is, the co-adsorption of OH and H, is much stronger than the molecular H2O adsorption. The energy barrier of H2O dissociation is 137.58 kJ mol−1 on a clean Al(111) surface, however, it decreases to 38.18 kJ mol−1 with the aid of a pre-adsorbed O atom, suggesting that a pre-adsorbed O atom can promote the dehydrogenation reaction of H2O.
1. Introduction
Understanding the interaction of gas molecules (O2 and H2O) with metal surfaces is critically important for many applications such as corrosion. Aluminum and its alloys are widely used outdoors1–6 because of their excellent mechanical properties and corrosion resistance. It has been shown that corrosion products play an important role in their corrosion resistance.7 The adsorption and reaction of O2 and H2O on metal surfaces is crucial to the formation of oxide films. Because of the absence of d-electrons and a simple geometrical structure, a gas/Al system is simpler than transition metals. As mentioned by Leygraf and Graedel,8 the time scale for surface film formation is approximately one microsecond, which means it occurs too fast to be observed or detected by experiments. Thus, theoretical calculations must be used to investigate the mechanism of the gas adsorption.The oxidation process is a key step of dissociative adsorption of O2 on the surface. To gain some insights into the mechanism of the oxidation of Al surfaces, O2 adsorption on Al(111) surfaces has been studied experimentally and theoretically by researchers.9–18 Pashutski et al.9 studied the adsorption of O2 on Al(100) at 80 K using Auger and X-ray photoelectron spectroscopy. The results showed that AlxOy oxides were formed in x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y ratios from 3:1 to 1:1 at low coverage, and the oxide layer transformed to the familiar Al2O3 at higher coverage or upon heating to room temperature. Experiments clearly indicate a mysteriously large number of adsorbed single oxygen atoms instead of pairs.10,11 The calculation results show that O2 can adsorb on multiple original sites on the Al(111) surface.12,13 Experiments and theoretical calculations support chemisorption only on the fcc site of the first layer of Al(111),14,15 and O2 does not penetrate into subsurface sites.16 Liu et al.17 predicted that O2 molecule can be adsorbed on the Al(111) surface with a barrier of approximately 0.2–0.4 eV, and the lowest unoccupied molecular orbital of O2 is higher than the Fermi level of the Al(111) surface, which is responsible for the barrier of the O2 adsorption. Florian Libisch et al.18 also investigated the origin of the energy barrier for chemical reactions of O2 on Al(111). The results show that correct barriers arise naturally when embedded correlated electron wave functions are used to capture the physics of the interaction of O2 with the metal surface. They suggested that the barrier originates from an abrupt charge transfer.
:y ratios from 3:1 to 1:1 at low coverage, and the oxide layer transformed to the familiar Al2O3 at higher coverage or upon heating to room temperature. Experiments clearly indicate a mysteriously large number of adsorbed single oxygen atoms instead of pairs.10,11 The calculation results show that O2 can adsorb on multiple original sites on the Al(111) surface.12,13 Experiments and theoretical calculations support chemisorption only on the fcc site of the first layer of Al(111),14,15 and O2 does not penetrate into subsurface sites.16 Liu et al.17 predicted that O2 molecule can be adsorbed on the Al(111) surface with a barrier of approximately 0.2–0.4 eV, and the lowest unoccupied molecular orbital of O2 is higher than the Fermi level of the Al(111) surface, which is responsible for the barrier of the O2 adsorption. Florian Libisch et al.18 also investigated the origin of the energy barrier for chemical reactions of O2 on Al(111). The results show that correct barriers arise naturally when embedded correlated electron wave functions are used to capture the physics of the interaction of O2 with the metal surface. They suggested that the barrier originates from an abrupt charge transfer.
There are also many papers that have investigated the adsorption and desorption kinetics of water on aluminum,19,20 the geometric, electronic and vibrational structure of the adsorbed layer,19,21,22 and the oxidation kinetics of aluminum with water.23,24 The adsorbed form of H2O is predominantly molecular on the clean surface at low temperature, and in the presence of oxygen, the adsorbed form is predominantly dissociated. The production of adsorbed hydroxyl species from water reaches a maximum at 250 K on the clean surface and 350 K on a surface with pre-adsorbed O atoms.25 A study by Paul and Hoffman showed that H2O decomposed preferentially to surface-bound hydroxyl species. They also found that H2O reversibly adsorbed on the Al(100) surface via hydrogen bonding, or H2O dissociated into H and OH species.26 Guo et al.27 studied the energy barriers for the water dissociation processes using the nudge elastic band method. The results showed that hydrogen atom dissociation from H2O requires 248.32 kJ mol−1 of energy on a clean Al(111) surface, whereas the dissociating energy decreased to 128.53 kJ mol−1 with the aid of O adsorption.
An accurate atomistic description of the H2O-solid interface is crucial for understanding the oxidation mechanism of Al. Although scanning probe techniques, specifically scanning tunneling microscope, have contributed significantly to the field by providing detailed insight into the structure and dynamics of H2O-adsorbed structures at the nanoscale, such studies are limited to well-defined, single crystal metal surfaces at low temperature and under ultra-high vacuum conditions. Computer calculation techniques, specifically density functional theory (DFT), have played a central role in understanding the mechanism of the interaction of H2O with metal surfaces,28 whereas these calculation remain an important problem. This is mainly because the standard DFT fails to describe the non-local van der Waals (vdW) dispersion forces, which are related to H2O adsorption and weak adsorption systems in general.28,29 As the problem exposed, a number of developments with DFT based schemes for dealing with vdW dispersion forces have been proposed.30 The H2O adsorption structures have been considered using dispersion-corrected DFT.31–34 Indeed, the studies have indicated that vdW dispersion forces should be accounted for when describing the interaction between a H2O molecule and a metal surface.
Here, we report a standard DFT study of the molecular and dissociative adsorptions of O2 on a clean Al(111) surface. We use a vdW-DFT method to investigate the molecular adsorption of the H2O on clean and oxygen pre-adsorbed Al(111) surfaces. The dissociation adsorption of H2O molecule, that is, the co-adsorption of OH and H, was calculated using standard DFT after dispersion force testing. We analyze the structural evolutions, adsorption energies, charge transfer and partial density of states (PDOS) of the adsorbed structures and discuss the impact of the pre-adsorbed O atom on the H2O adsorption behavior.
2. Computational details
2.1 Adsorption calculation
All calculations presented in this work were conducted using MedeA-VASP 5.4 software,35,36 which is a fast and highly reliable electronic structure method that is based on DFT.37 The calculation was conducted in a plane-wave basis, using the projector-augmented wave method.38 The exchange–correlation functional for describing the interactions was GGA-PBE.39 The adsorption calculations were conducted on 6-layer slabs of Al(111) with a 12 Å vacuum gap. A (3 × 3) mesh was used for the adsorption calculation. The adsorbates and the three uppermost surface layers were allowed to move freely, and the bottom three layers were fixed. The electronic iterations convergence was 10−5 eV using the Normal (blocked Davidson) algorithm. Periodic boundary conditions were set, leading to an infinite periodic system.The adsorption energies (Ead) were calculated from the following expression:
| Ead = Eads + Esub − Eads/sub | (1) |
The surface energy (γsurf) was calculated from the following expression:
| γsurf = (Esub − nEatom-bulk)/2A | (2) |
All calculations involving O2 were performed with a spin polarization to adequately describe the triplet state of O2. Tests were established when the vdW dispersion force was considered for the molecular and dissociated adsorption of H2O on the clean and O pre-adsorbed Al(111) surface. Based on previous calculations,40 accounting for vdW dispersion forces does not change the adsorption structures. Table 1 shows the change of adsorption energy before and after considering non-local vdW dispersion forces. Comparing the DFT calculation with the vdW-DFT calculation in terms of adsorption energies, the vdW dispersion forces have a significant effect on the molecular adsorption of H2O on the clean and oxygen pre-adsorbed Al(111) surface. Therefore, the vdW-DFT approximately takes into account the dispersive forces and vdW interactions in the molecular adsorption of H2O. The optimized vdW functional based on the Becke 86 (optB86-vdW41,42), was chosen as the exchange functional, which tends to exhibit the smallest errors for most of the systems investigated. The non-local vdW correlation was not defined for a spin polarized system. The slab models were calculated using a (4 × 4 × 1) Monkhorst–Pack grid.43 All calculations were performed using a 520 eV cut off energy. The post-processing of the results, which was based on structure and charge density, was constructed using VESTA.44
| Adsorbed structure | Adsorption energy (eV) | ΔE/Eads-DFT (%) | |
|---|---|---|---|
| DFT | vdW-DFT | ||
| H2O(top) | 0.224 | 0.398 | 77.7 |
| O(fcc)–H2O(top)–O(fcc) | 0.454 | 0.668 | 47.1 |
| OH(fcc)–H(top) | 7.539 | 7.728 | 2.5 |
| OH(fcc)–OH(fcc)–O(fcc) | 6.859 | 6.979 | 1.7 |
The so-called Bader scheme for dividing the slab surface into atomic regions has been used,45 which proved to be robust and efficient. Included in MedeA-VASP as a property module, this approach is based on a classic algorithm, which was proposed by Bader and, implemented by Henkelman et al.46 The calculation of difference charge density between the self-consistent pseudo charge density and the superposition of atomic charge densities is one of the most significant applications of pseudopotential theory. The property of PDOS was calculated to investigate the interaction between different atoms. From the PDOS diagram, the peak shifting indicates that the electronic number of the atom within a certain energy interval changed. The interaction between different atoms may relate to the overlapping of peaks.
2.2 Transition states search calculations
For the transition states search (TSS) calculation, the Nudged elastic band47 method was used to map the minimum energy path between the initial system and the final system at a spring constant of 5 eV Å−2. The initial images were created from linear interpolation. Reaction coordinate is the normalized coefficient of linear interpolation. Transition states (TS) were searched for the highest saddle point only. Optimization of transition states was attempted. The image closest to a saddle point was allowed to climb up into the saddle point if the largest force on an atom was smaller than 0.5 eV Å−1. The SCF of each iteration was started from wave functions of the previous iteration. The convergence was 0.05 eV Å−1. Diagonal elements of the inverse Hessian were initially set to 0.01 Å2 eV−1. The TSS calculation used normal precision and a plane wave cutoff energy of 520 eV. The electronic iterations convergence was 10−5 eV using the Normal (blocked Davidson) algorithm and reciprocal space projection operators. The K point was 4 × 4 × 1. The vdW dispersion correction is considered for the TSS of the dissociation of H2O.3. Results and discussion
3.1 O atoms adsorption on the clean Al(111) surface
The calculated surface energy and work function of a clean six-layer Al(111) surface are 1.06 J m−2 and 4.12 eV, respectively, which agree well with corresponding experimental values of 1.14 J m−2 (ref. 48) and 4.24 eV,49 respectively. Four highly symmetric adsorption sites were considered: top site, bridge site, fcc site and hcp site, as shown in Fig. 1. The O atoms could only adsorb on three hollow sites which are the fcc site and hcp site at coverages of 1/9 and 2/9 ML in our study. The adsorbed structures are listed in Table 2 and marked in Fig. 1. The geometrical parameters, adsorption energies and charge transfer details are provided in Table 2. For the single O atom adsorption, O(fcc1) and O(hcp1) structures were investigated. A chemical adsorption was observed, which was due to the close distance of Al–O (1.87 Å) and which is smaller than the sum of their ionic radii. The adsorption energy of the O(fcc1) structure is larger than that of O(hcp1) indicating a more stable adsorption, whereas the charge prefers to transfer to an O atom adsorbed on a hcp site. For the O adsorption on surface at coverages of 2/9 ML, six adsorbed structures were investigated, in which the O atoms were adsorbed on the fcc site and hcp sites at the nearest and next nearest adjacent distances. The adsorbed structures, geometrical parameters, adsorption energies and charge transfer details are shown in Table 2. The adsorption energies were calculated as the single O atom, which are basically consistent with previous calculations.50 O(fcc1)–O(fcc2) is the most stable adsorbed structure of all the calculated structures at coverages of 2/9 ML according to the adsorption energies.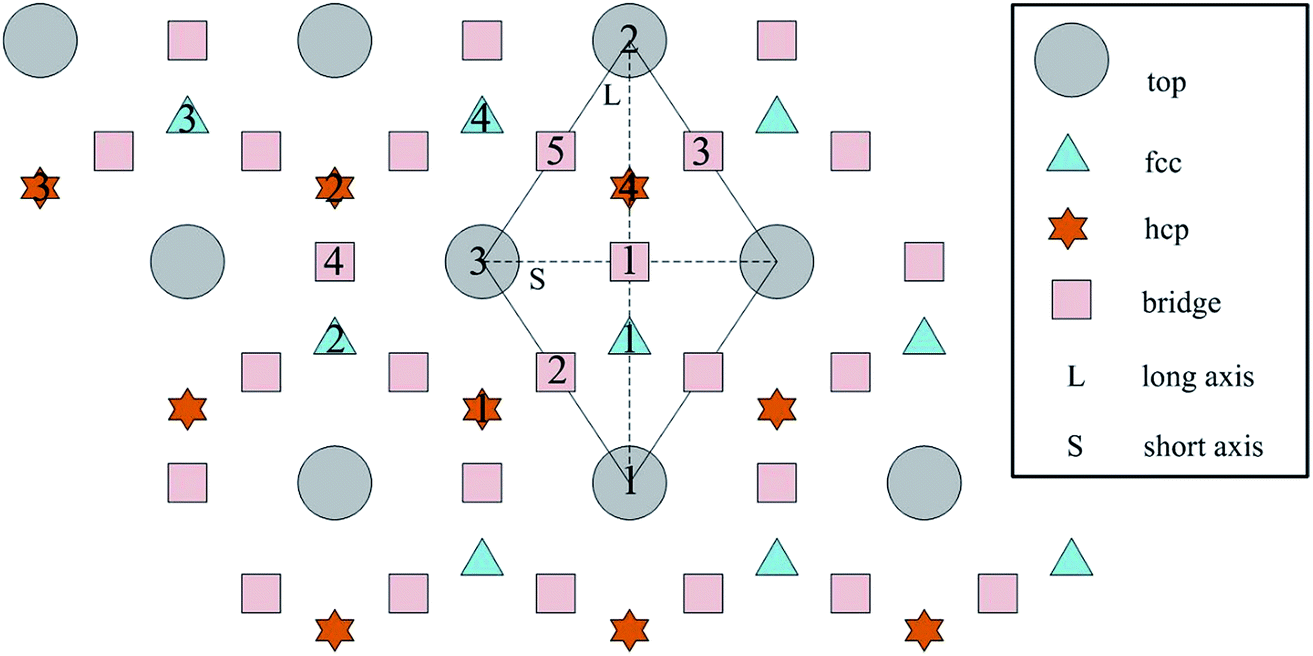 | ||
| Fig. 1 The high symmetrical adsorption sites on the Al(111) surface. | ||
| Adsorbed structure | Distance (Å) | Adsorption energy (eV) | Charge transfer | |||
|---|---|---|---|---|---|---|
| Al–O(fcc) | Al–O(hcp) | O–O | Al → O(fcc) | Al → O(hcp) | ||
| O(fcc1) | 1.87 | — | — | 7.52 | 1.730 | — |
| O(hcp) | — | 1.87 | — | 7.21 | — | 1.759 |
| O(fcc1)–O(fcc2) | 1.86(1.86) | — | 2.88 | 7.61 | 1.723(1.721) | — |
| O(fcc1)–O(fcc3) | 1.86(1.87) | — | 4.96 | 7.49 | 1.737(1.731) | — |
| O(fcc1)–O(hcp1) | 1.85 | 1.86 | 2.39 | 7.22 | 1.686 | 1.711 |
| O(fcc1)–O(hcp2) | 186 | 1.87 | 3.32 | 7.40 | 1.734 | 1.763 |
| O(hcp1)–O(hcp2) | — | 1.86(1.87) | 2.87 | 7.30 | — | 1.745(1.744) |
| O(hcp1)–O(hcp3) | — | 1.87(1.86) | 4.96 | 7.14 | — | 1.753(1.771) |
To further understand the mechanism of the O atom adsorption on the Al(111) surfaces, TSS calculations of the O atom along Al(111) surface and O pre-adsorbed Al(111) surface were calculated. The fcc site is the most stable site, and hcp is the metastable site of single O atom on the clean Al(111) surface. The hcp–fcc diffusion pathways of O atom on the clean and O(fcc) pre-adsorbed Al(111) surfaces were calculated. As shown in Fig. 2 and 3, energy barrier of 25.91 kJ mol−1 for the O diffusion on the clean surface is lightly lower than the corresponding values of 28.67 kJ mol−1 for the O diffusion on the O(fcc) pre-adsorbed Al(111) surface, suggesting that the adsorbed O atom on the fcc site slightly inhibits the diffusion of O atom on the Al(111) surface. The initial and final structures and their TS structures are also shown in Fig. 2 and 3, respectively. The bridge site is the adsorption site of the TS for both the clean surface and for the O(fcc) pre-adsorbed Al(111) surface.
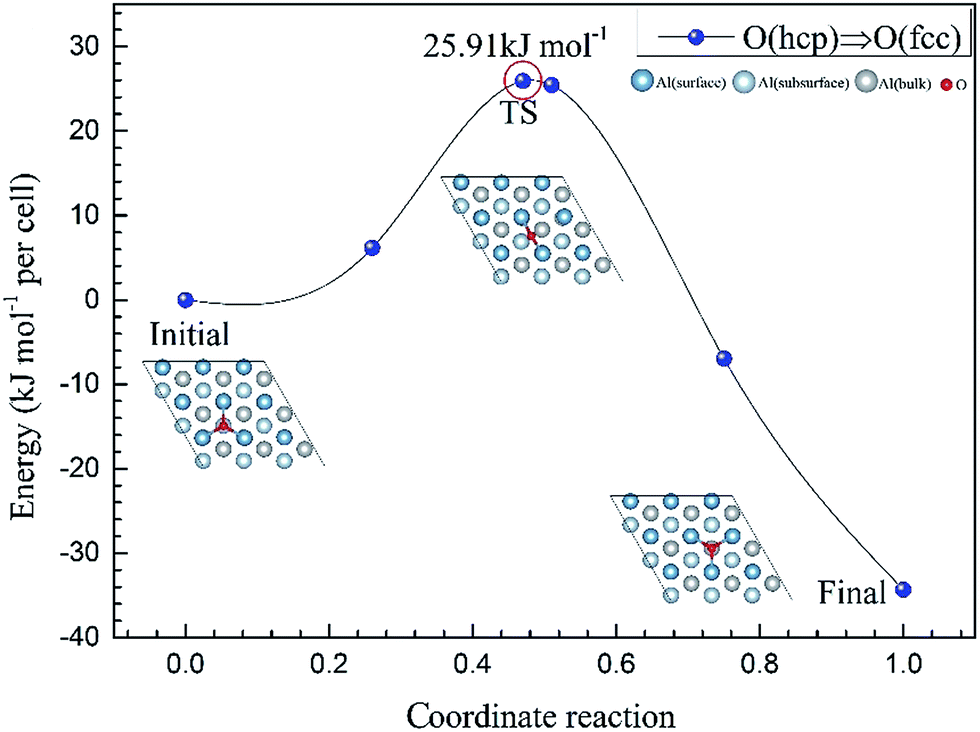 | ||
| Fig. 2 The TSS results of single O atom along O(hcp)–O(fcc) pathway on the clean Al(111) surface. | ||
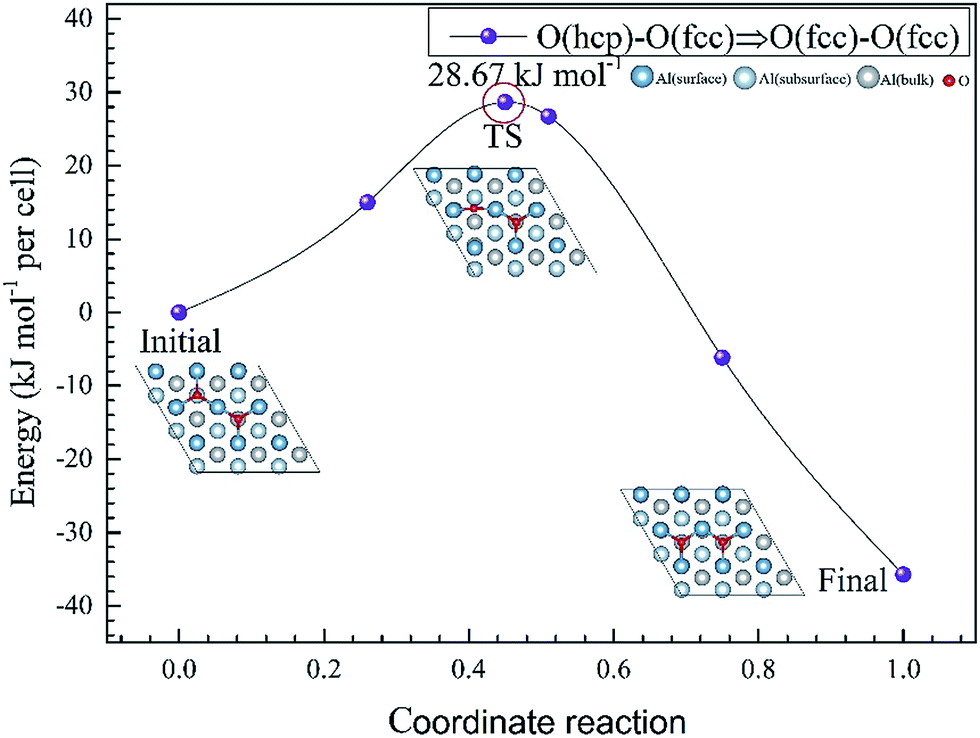 | ||
| Fig. 3 The TSS results of single O atom along O(hcp)–O(fcc) pathway on the O(fcc) pre-adsorbed Al(111) surface. | ||
3.2 Molecular adsorption of H2O on Al(111) surfaces
| Adsorbed structure | Top view | Side view |
|---|---|---|
| H2O(top)–S |
|
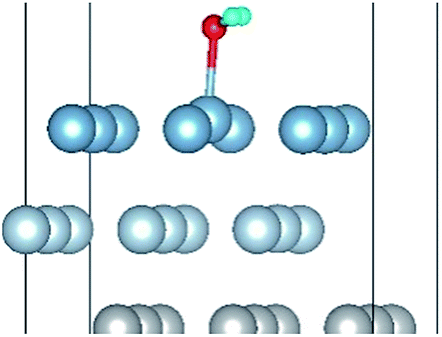
|
| H2O(top)–L |
|
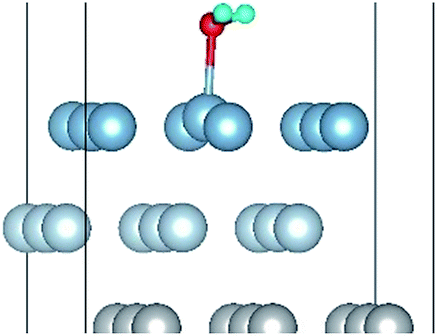
|
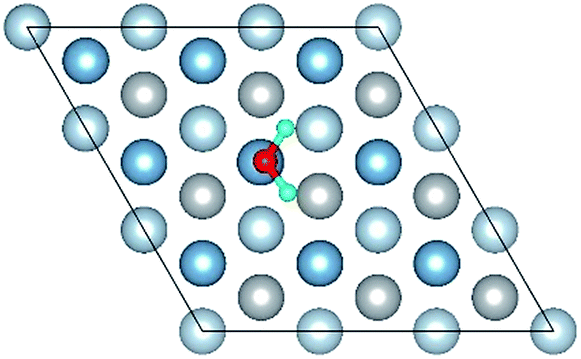
| O(fcc)–H2O(top)–O(fcc)–S |
|
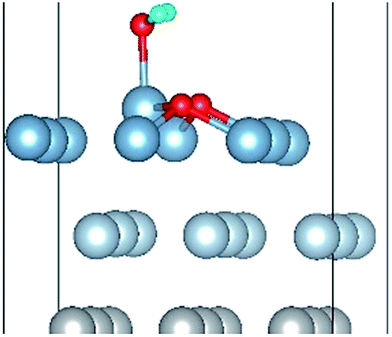
|
| O(fcc)–H2O(top)–O(fcc)–L |
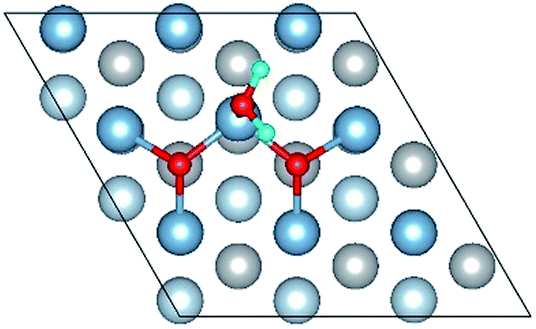
|
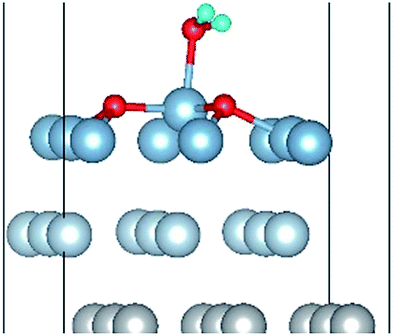
|
| Remarks |

|
|
| Adsorbed structure | Distance (Å) | ∠HOH (°) | Adsorption energy (eV) | Charge transfer | |
|---|---|---|---|---|---|
| Al–O | H–O | ||||
| H2O(top3)–S | 2.15 | 0.98 | 106.2 | 0.398 | 0.079 |
| H2O(top3)–L | 2.15 | 0.98 | 106.4 | 0.398 | 0.085 |
| O(fcc1)–H2O(top3)–O(fcc2)–S | 2.04 | 0.98 | 106.7 | 0.668 | 0.004 |
| O(fcc1)–H2O(top3)–O(fcc2)–L | 2.03 | 0.98 | 107.0 | 0.677 | 0.032 |
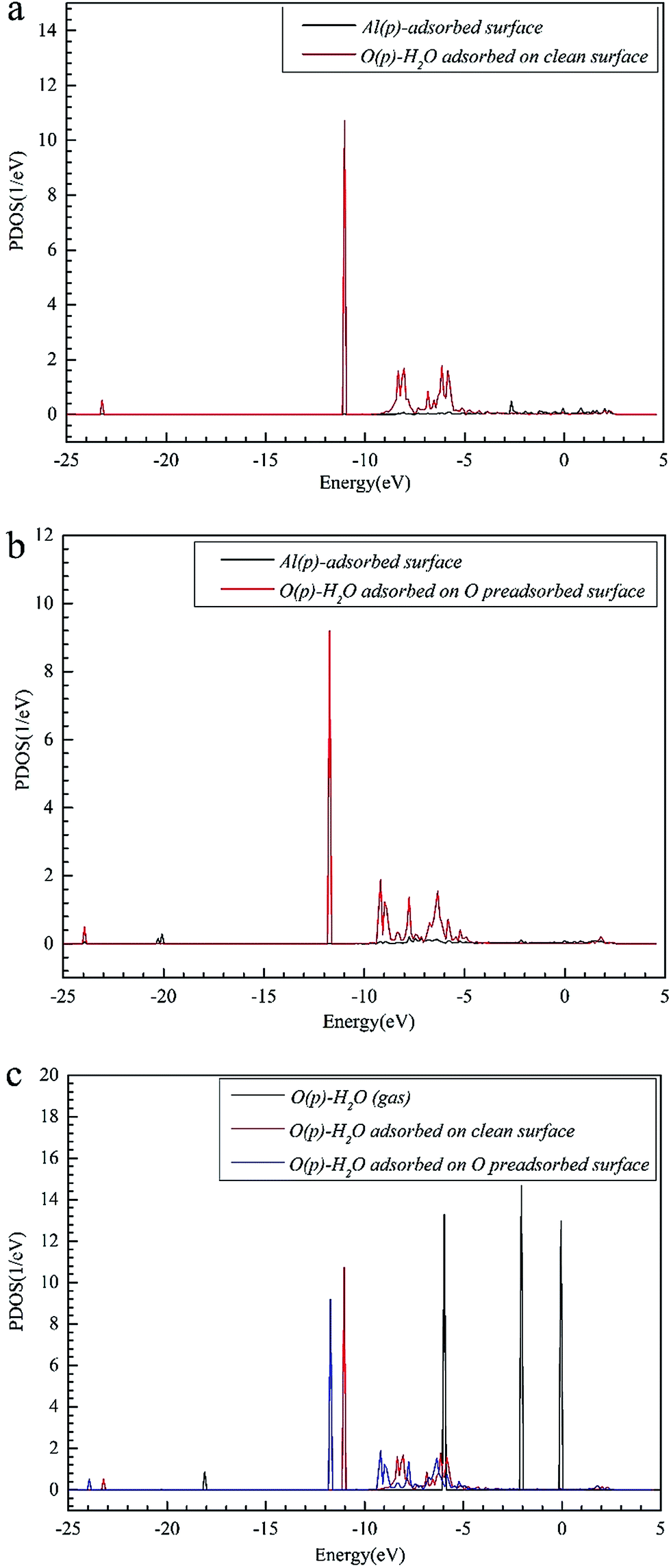 | ||
| Fig. 4 The PDOS results of (a) Al(p) of the clean Al(111) surface and O(p) of H2O adsorbed on the clean Al(111) surface; (b) Al(p) of the clean Al(111) surface and O(p) of H2O adsorbed on the O pre-adsorbed Al(111) surface; (c) O(p) of H2O in gas, H2O adsorbed on the clean Al(111) surface, and H2O adsorbed on the O pre-adsorbed surface. | ||
The closer distance of Al–O and larger adsorption energies indicate a stronger adsorption for the H2O on the O pre-adsorbed surface. The reduced charge transfer occurs because some of the electrons on the surface are controlled by the pre-adsorbed O atoms, which have a high electronegativity. The PDOS of Al(p) of surface and O(p) of the H2O molecule was calculated for the O(fcc1)–H2O(top3)–O(fcc2)–S structure shown in Fig. 4b. Similarly, the resonance of the PDOS peaks of O(p) and Al(p) of O(fcc1)–H2O(top3)–O(fcc2)–S adsorbed structure occurs over the entire energy range. Fig. 4c shows the PDOS of O(p) of H2O in gas, H2O adsorbed on the clean surface and H2O adsorbed on the O pre-adsorbed surface. The downshift of the O(p) state with respect to the gas is characterized by the displacement of the peak. The results illustrate that the adsorption of H2O on the O pre-adsorbed surface is stronger adsorption on the clean surface, which agrees well with the conclusion inferred from geometrical parameters and adsorption results.
3.3 The dissociated adsorption of H2O on Al(111)
We discuss the dissociated adsorption behavior of H2O, that is, the co-adsorption of H and OH on the clean and O pre-adsorbed Al(111) surfaces. In total 17 initial and final structures of OH and H co-adsorbed on the clean Al(111) surface were investigated, which are listed in Table 5. The initial and final structures were characterized as shown in Fig. 1. The H atom can be adsorbed on the top2, top3, fcc2, and bridge1 sites when OH adsorbs on the top1 site and it migrates from bridge4 to fcc2 and hcp4 to bridge 1 sites, respectively. For the adsorption of OH on the fcc1 site, the H atom can adsorb on top2, and fcc2 directly and migrates from hcp4 to top3, and bridge4 to fcc2. The OH will migrate to the nearest bridge site when its initial site is hcp corresponding to H atom of different adsorbed sites. The geometrical parameters, adsorption energies and charge transfer details of OH and H co-adsorbed structures on clean Al(111) surface is shown in Table 5. There are chemical bonds between HO–Al and H–Al according to their distance and all structures have larger adsorption energies. The charge transfer from the surface to OH and H is much larger than the adsorption of H2O molecule. Therefore, offering enough charge to H2O is one of the necessary conditions to cause it to dissociate. Table 6 shows three possible adsorbed structures of OH and O on O(fcc1)–O(fcc2) pre-adsorbed Al(111) surfaces. The H atom that dissociated from H2O bonds with one of two pre-adsorbed O atoms and adsorb on the surface in the form of OH. The geometrical parameters, adsorption energies and charge transfer details are shown in Table 7. Comparing with the adsorption of OH and H on the clean surface, both the adsorption energies and charge transfer of the adsorption on O pre-adsorbed surface decrease. Thus, the dissociated H2O prefers to adsorb on a clean surface rather than on the O pre-adsorb surface, whereas the required charge transferred to H2O that adsorbed on O pre-adsorbed surface was significantly reduced.| Initial structures | Adsorbed structures | Distance (Å) | E ads (eV) | Charge transfer | ||
|---|---|---|---|---|---|---|
| Al–O | Al–H | H–O | ||||
| OH(top1)–H(top2) | OH(top1)–H(top2)–L | 1.74 | 1.62 | 0.97 | 7.14 | 1.412 |
| OH(top1)–H(top3) | OH(top1)–H(top3)–S | 1.74 | 1.62 | 0.97 | 7.12 | 1.421 |
| OH(top1)–H(fcc2) | OH(top1)–H(fcc2) | 1.75 | 1.92 | 0.97 | 7.28 | 1.705 |
| OH(top1)–H(bridge4) | OH(top1)–H(fcc2) | 1.75 | 1.92 | 0.97 | 7.28 | 1.701 |
| OH(top1)–H(hcp4) | OH(top1)–H(bridge1) | 1.73 | 1.81 | 0.98 | 7.19 | 1.689 |
| OH(fcc1)–H(top2) | OH(fcc1)–H(top2) | 2.07 | 1.63 | 0.98 | 7.33 | 1.638 |
| OH(fcc1)–H(fcc2) | OH(fcc1)–H(fcc2) | 2.07 | 1.92 | 0.98 | 7.45 | 1.891 |
| OH(fcc1)–H(hcp4) | OH(fcc1)–H(top3) | 2.05 | 1.62 | 0.98 | 7.39 | 1.636 |
| OH(fcc1)–H(bridge4) | OH(fcc1)–H(fcc2) | 2.07 | 1.63 | 0.98 | 7.45 | 1.890 |
| OH(hcp4)–H(top1) | OH(bridge1)–H(top1) | 1.90 | 1.66 | 1.01 | 7.54 | 1.629 |
| OH(hcp4)–H(fcc2) | OH(bridge1)–H(fcc2) | 1.94 | 1.91 | 0.98 | 7.44 | 1.854 |
| OH(hcp4)–H(hcp1) | OH(bridge1)–H(hcp1) | 1.94 | 1.91 | 0.98 | 7.25 | 1.868 |
| OH(hcp4)–H(bridge4) | OH(bridge5)–H(bridge4) | 1.93 | 1.74 | 0.98 | 7.34 | 1.783 |
| OH(bridge2)–H(top2) | OH(bridge2)–H(top2) | 1.94 | 1.62 | 0.98 | 7.31 | 1.570 |
| OH(bridge2)–H(fcc4) | OH(bridge2)–H(fcc4) | 1.95 | 1.92 | 0.98 | 7.45 | 1.846 |
| OH(bridge2)–H(hcp4) | OH(bridge2)–H(bridge3) | 1.93 | 1.83 | 0.98 | 7.41 | 1.756 |
| OH(bridge2)–H(bridge5) | OH(bridge2)–H(fcc4) | 1.94 | 1.92 | 0.98 | 7.45 | 1.851 |
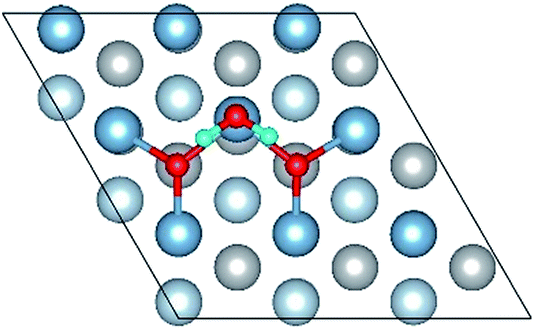
| Adsorbed structure | Top view | Side view |
|---|---|---|
| OH(bridge1)–OH(top2)–O(fcc2) |
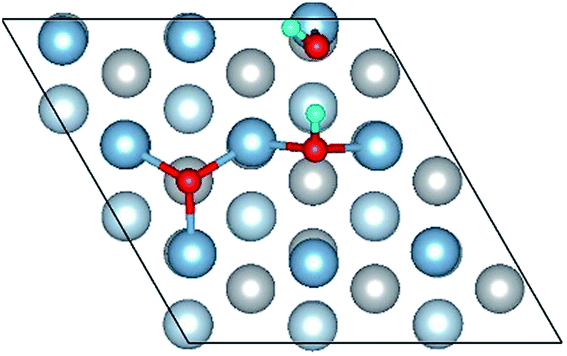
|
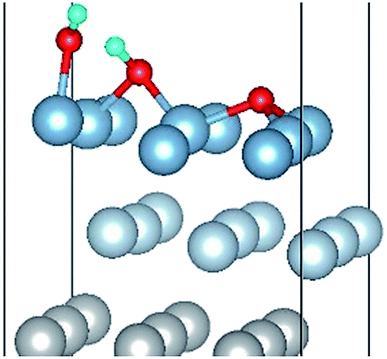
|
| OH(fcc1)–OH(fcc4)–O(fcc2) |
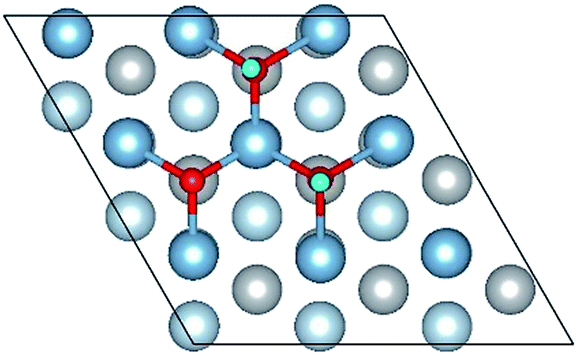
|
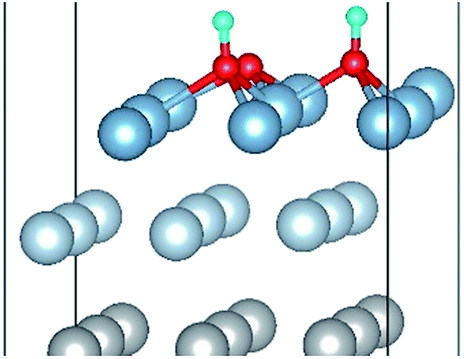
|
| OH(bridge2)–OH(bridge5)–O(fcc2) |
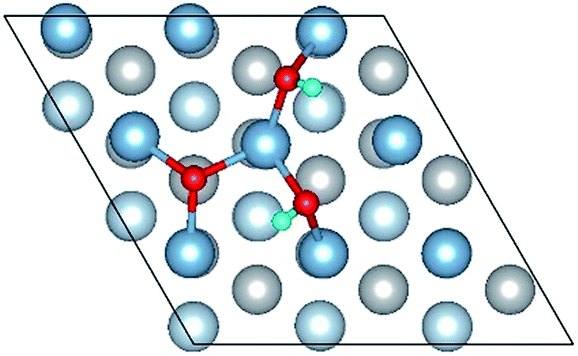
|
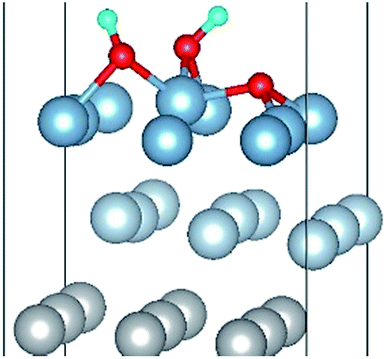
|
| Remarks |

|
|
| Adsorbed structure | Distance (Å) | E ads (eV) | Charge transfer | ||
|---|---|---|---|---|---|
| Al–O(fcc) | Al–O(OH) | O–H | |||
| OH(bridge1)–OH(top2)–O(fcc2) | 1.86 | 1.86 | 1.00 | 6.87 | 0.330 |
| OH(fcc1)–OH(fcc4)–O(fcc2) | 1.87 | 2.06 | 0.98 | 6.85 | 0.375 |
| OH(bridge2)–OH(bridge5)–O(fcc2) | 1.86 | 1.92 | 0.98 | 6.76 | 0.311 |
To elucidate the mechanisms of the dehydrogenated reaction of single H2O on the clean and O pre-adsorbed surfaces, the energy barriers for the H2O dissociation processes were calculated using the TSS method. The energy barriers and structural evolution of the systems for H2O dissociation on the clean and O pre-adsorbed Al(111) surfaces are shown in Fig. 5 and 6. There is a high energy barrier for H2O dissociation on the clean surface. For the case of H2O dissociation on the O pre-adsorbed surface, the dissociation energy barrier reduced significantly compared with that on clean surface, which indicates that the active energy of H2O dissociation decreased due to the pre-adsorbed O atom. Upon comparison of the energy of the initial and final adsorbed structures, the dissociation of H2O on the clean surface was found to be spontaneous reaction with a high energy barrier. However, the dissociation of H2O on the O pre-adsorbed surface is a non-spontaneous process with a low active energy barrier. The pre-adsorbed O is an electronegative atom that makes it more attractive than Al for the H atom of H2O. When O adsorbs on the Al(111) surface, the electrons of Al are attracted toward the O atom. Therefore, the pre-adsorbed O will weaken the O–H bond and cause it easily dissociate.
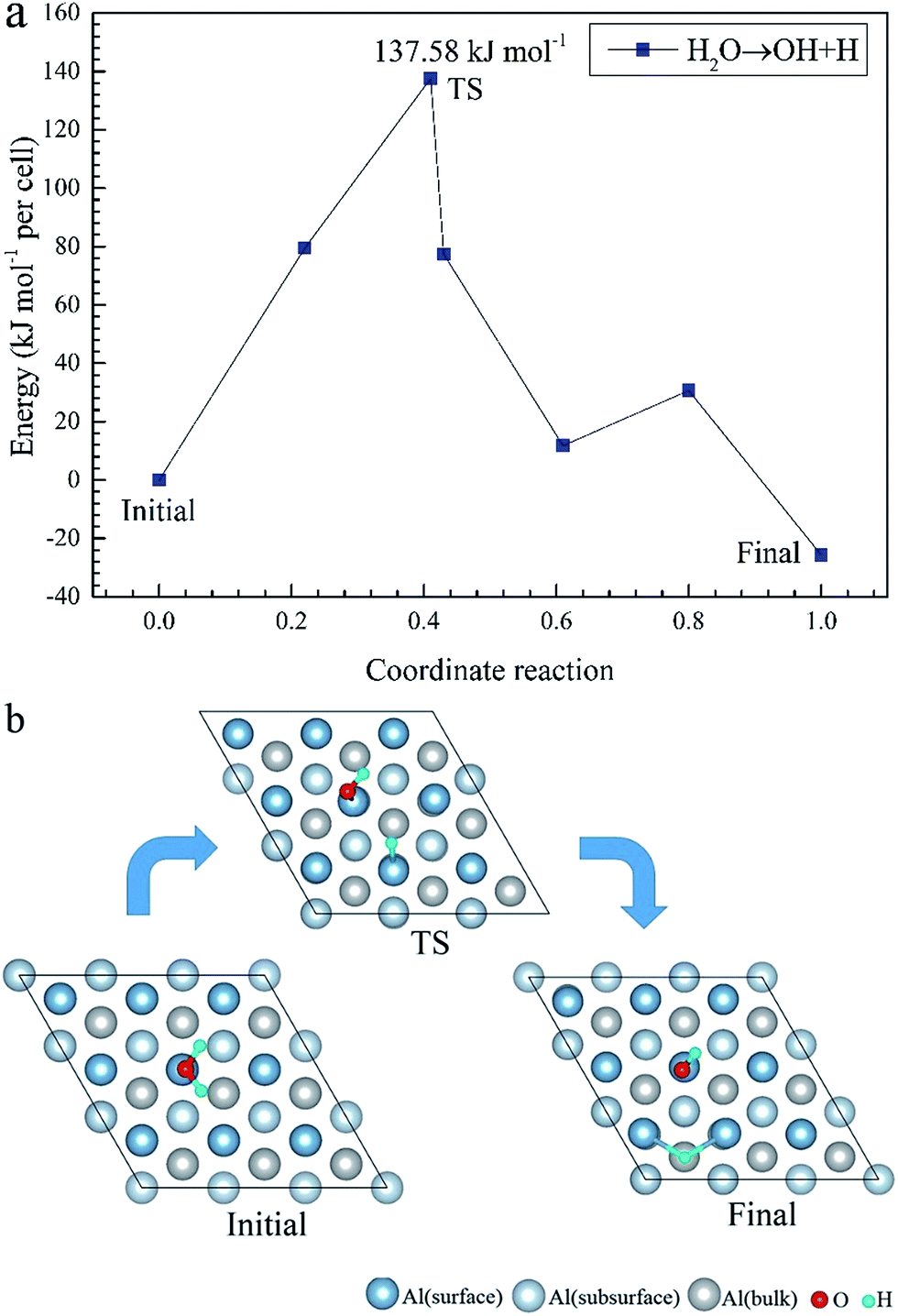 | ||
| Fig. 5 The TSS results of dissociated reaction of H2O molecule on the clean Al(111) surface. | ||
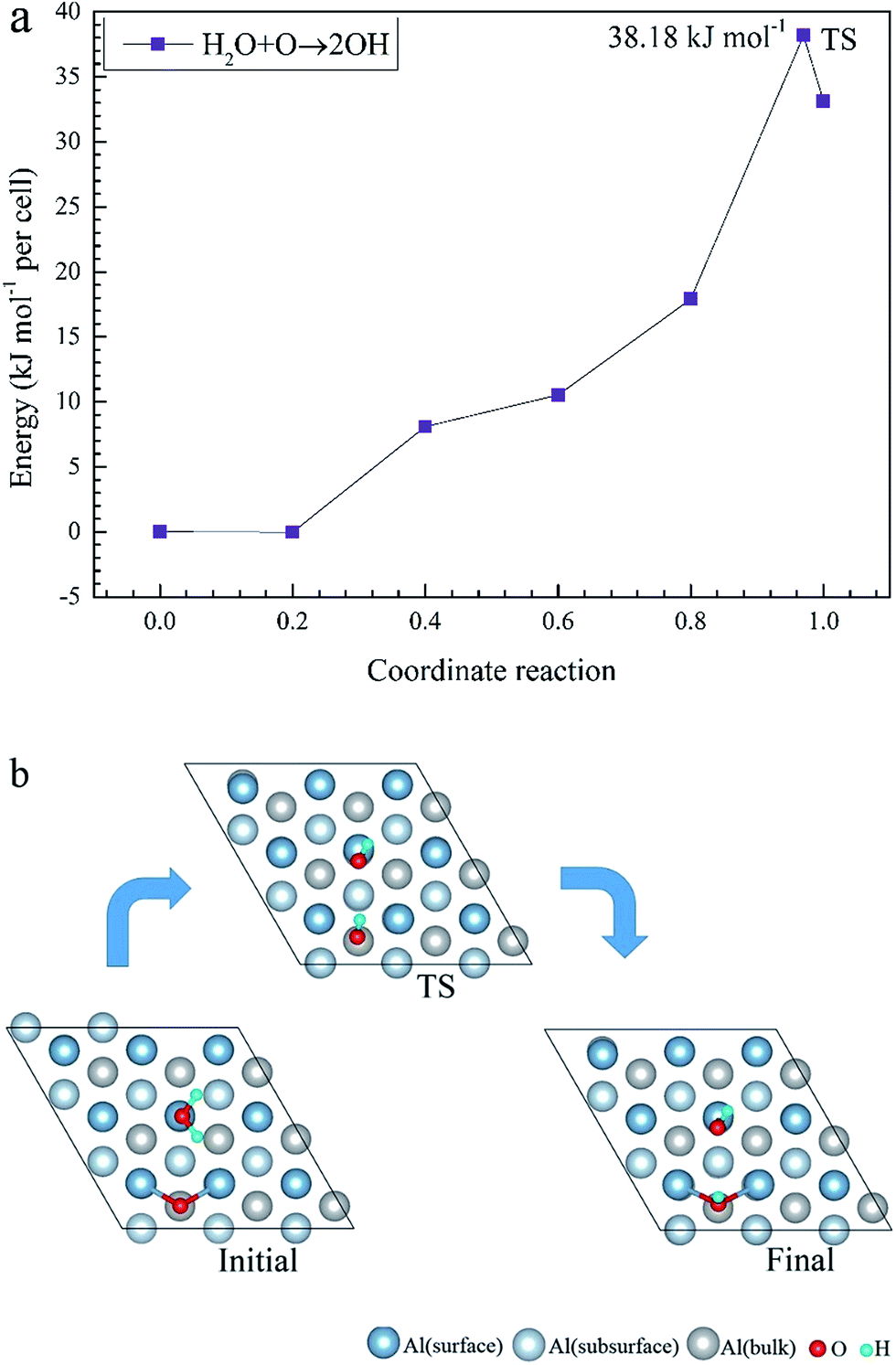 | ||
| Fig. 6 The TSS results of dissociated reaction of H2O molecule on the O(fcc) pre-adsorbed Al(111) surface. | ||
4. Conclusions
Using first-principles calculations that are based on DFT, the molecular and dissociative adsorptions of O2 and H2O on clean and oxygen pre-adsorbed Al(111) surfaces were investigated. The adsorbed structures, adsorption energy, PDOS, charge transfer from surface to the adsorbates and energy barriers for O atom migration and H2O dissociation are calculated. The main conclusions are summarized as follows:(1) For the adsorption of O2 on the clean Al(111) surface, O2 dissociates into O atoms which can adsorb on the fcc and hcp sites of the clean Al(111) surface, and the ranking of adsorption site stability was found to be fcc > hcp. The energy barriers for a single O atom along the hcp–bridge–fcc pathway on the clean and O(fcc) pre-adsorbed Al(111) surface are 25.91 kJ mol−1 and 28.67 kJ mol−1 indicating that the pre-adsorbed O atom has a weak inhibiting effect on O atom migration.
(2) H2O can only adsorb in the form of a molecule on both the clean and O pre-adsorbed Al(111) surfaces, and the adsorption is very weak. The PDOS results show that the pre-adsorbed O atoms on the surface can strengthen the adsorption of H2O and promote its deformation.
(3) The dissociation adsorption of H2O, that is, the co-adsorption of OH and H, is much stronger than the molecular adsorption of H2O. On the clean Al(111) surface, the dissociation of a single H atom from H2O requires 137.58 kJ mol−1. However, with the aid of the pre-adsorbed O atom, the dissociating energy decreases to 38.18 kJ mol−1, suggesting that pre-adsorbed O can promote the dissociation of H2O.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 51222106), the Fundamental Research Funds for the Central Universities (No. 230201306500002) and National Basic Research Program of China (973 Program) (No. 2014CB643300).References
- B. B. Wang, Z. Y. Wang, W. Han and W. Ke, Corros. Sci., 2012, 55, 63–70 CrossRef.
- S. Sun, Q. Zheng, D. Li and J. Wen, Corros. Sci., 2009, 51, 719–727 CrossRef CAS.
- Y. Shi, Z. Zhang, J. Su, F. Cao and J. Zhang, Corros. Sci., 2006, 51, 4977–4986 CAS.
- S. Sun, Q. Zheng, D. Li, S. Hu and J. Wen, Corros. Sci., 2011, 53, 2527–2538 CrossRef CAS.
- Y. L. Cheng, Z. Zhang, F. H. Cao, J. F. Li, J. Q. Zhang, J. M. Wang and C. N. Cao, Corros. Sci., 2004, 46, 1649–1667 CrossRef CAS.
- N. D. Alexopoulos, Corros. Sci., 2012, 55, 289–300 CrossRef CAS.
- S. Liu, H. Sun, L. Sun and H. Fan, Corros. Sci., 2012, 65, 520–527 CrossRef CAS.
- C. Leygraf and T. E. Graedel, Atmospheric corrosion, electrochemical society series, Wiley-interscience, 2000 Search PubMed.
- A. Pashutski, A. Hoffman and M. Folman, Surf. Sci. Lett., 1989, 208, L91–L97 CrossRef CAS.
- A. J. Komrowski, J. Z. Sexton, A. C. Kummel, M. Binetti, O. Weiße and E. Hasselbrink, Phys. Rev. Lett., 2001, 87, 246103 CrossRef CAS PubMed.
- H. Brune, J. Wintterlin, R. J. Behm and G. Ertl, Phys. Rev. Lett., 1992, 68, 624 CrossRef CAS PubMed.
- X. Lei and L. Yang, Comp. Appl. Chem., 2011, 28, 793–796 CAS.
- D. Costa, T. Ribeiro and F. Mercuri, Adv. Mater. Interfaces, 2014, 1, 1300072 CrossRef.
- M. Shao, Y. Fu, R. Hu and C. Lin, Mater. Sci. Eng., A, 2003, 344, 323–327 CrossRef.
- X. Liu, G. S. Frankel, B. Zoofan and S. I. Rokhlin, Corros. Sci., 2007, 49, 139–148 CrossRef CAS.
- B. C. Mitrovic and D. J. O'Connor, Surf. Sci., 1998, 405, 261–270 CrossRef CAS.
- H.-R. Liu, H. Xiang and X. G. Gong, J. Chem. Phys., 2011, 135, 214702 CrossRef PubMed.
- J. Carrasco, J. Klimeš and A. Michaelides, Phys. Rev. Lett., 2013, 138, 024708 Search PubMed.
- F. P. Netzer and T. E. Madey, Surf. Sci., 1983, 127, L102–L109 CrossRef CAS.
- U. Memmert, S. J. Bushby and P. R. Norton, Surf. Sci., 1989, 219, 327–340 CrossRef CAS.
- J. E. Crowell, J. G. Chen, D. M. Hercules and J. T. Yates Jr, J. Chem. Phys., 1987, 86, 5804–5815 CrossRef CAS.
- P. B. Smith and S. L. Bernasek, J. Electron Spectrosc. Relat. Phenom., 1989, 49, 149–158 CrossRef CAS.
- F. Szalkowski, J. Chem. Phys., 1982, 77, 5224–5227 CrossRef CAS.
- H. D. Ebinger and J. T. Yates Jr, Surf. Sci., 1998, 412/413, 1–11 CrossRef CAS.
- J. E. Crowell, J. G. Chen, D. M. Hercules and J. T. Yates Jr, J. Chem. Phys., 1987, 86, 5804–5815 CrossRef CAS.
- J. Paul and F. M. Hoffman, J. Phys. Chem., 1986, 90, 5321–5324 CrossRef CAS.
- F. Y. Guo, C. G. Long, J. Zhang, Z. Zhang, C. H. Liu and K. Yu, Appl. Surf. Sci., 2015, 324, 584–589 CrossRef CAS.
- J. Carrasco, A. Hodgson and A. Michaelides, Nat. Mater., 2012, 11, 667–674 CrossRef CAS PubMed.
- A. Hodgson and S. Haq, Surf. Sci. Rep., 2009, 64, 381–451 CrossRef CAS.
- J. Klimeš and A. Michaelides, J. Chem. Phys., 2012, 137, 120901 CrossRef PubMed.
- I. Hamada, K. Lee and Y. Morikawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 115452 CrossRef.
- T. Kumagai, H. Okuyama, S. Hatta, T. Aruga and I. Hamada, J. Chem. Phys., 2011, 134, 024703 CrossRef CAS PubMed.
- A. Poissier, S. Ganeshan and M. V. Fernández-Serra, Phys. Chem. Chem. Phys., 2011, 13, 3375–3384 RSC.
- K. Tonigold and A. Gross, J. Comput. Chem., 2012, 33, 695–701 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. Lett., 1993, 47, 558–561 CAS.
- G. Kresse and J. Furthmüller, J. Phys. Chem. B, 1996, 54, 11169–11186 CAS.
- W. Kohn and L. J. Sham, Phys. Rev. A, 1965, 140, 1133–1138 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- F. A. Soria, P. Paredes-Olivera and E. M. Patrito, J. Phys. Chem. C, 2015, 119, 284–295 CAS.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- J. Klimeš, D. R. Bowler and A. Michaelides, J. Phys.: Condens. Matter, 2010, 22022201 Search PubMed.
- H. Monkhorst and J. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules-A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- J. Henkelman and H. J. Jonsson, Chem. Phys., 2000, 113, 9978–9985 Search PubMed.
- W. R. Tyson and W. A. Miller, Surf. Sci., 1977, 62, 267–276 CrossRef CAS.
- H. B. Michaelson, J. Appl. Phys., 1977, 48, 4729–4733 CrossRef CAS.
- A. Kiejna and B. I. Lundqvist, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 085405 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |