
A phosphotungstic acid-supported multifunctional organocatalyst containing 9-amino(9-deoxy)epi-cinchonidine and Brønsted acid and its application in asymmetric aldol reaction†
Ling Lan,
Guangxin Xie,
Tao Wu,
Dandan Feng and
Xuebing Ma*
Key Laboratory of Applied Chemistry of Chongqing Municipality, School of Chemistry and Chemical Engineering, Southwest University, Chongqing, 400715, P. R. China. E-mail: zcj123@swu.edu.cn; Fax: +86 2368253237; Tel: +86 23 68253237
First published on 6th June 2016
Abstract
A novel type of supported multifunctional organocatalyst CDNH2(n)–HPW combined 9-amino(9-deoxy)epi-cinchonidine (CDNH2) with Brønsted acid in the backbone of phosphotungstic acid was developed via facile precipitation of sodium tungstate with organophosphonic acid. The aldol addition of cyclohexanone to o, m or p-substituted benzaldehydes bearing electron-withdrawing substituents (–X and –NO2) afforded good yields (71–96%) and excellent stereoselectivities (anti/syn = 94/6–98/2, 87–97% ee). It was found that the arm length (n) and acid capacity had significant effects on the cooperative catalysis of CDNH2 and Brøsted acid in the aldol reaction. After the completion of the aldol reaction, the catalysts could be readily recovered in quantitative yield by centrifugation and possessed good reusability with no significant loss of catalytic performance in the five consecutive asymmetric aldol reactions.
Introduction
Natural cinchona alkaloid-derived primary amines, employed as homogeneous organocatalysts in asymmetric aldol,1 Michael,2 α-benzoyloxylation,3 α-alkylation,4 cycloaddition5 and cascade reactions,6 continue to act as powerful star organocatalysts able to effectively promote the chemical transformations of carbonyl compounds via enamine or iminium activation. As far as we know, to improve the economics and sustainability, the commonly used strategy by means of the heterogenization of these successful homogeneous organocatalysts by a covalent linkage to an organic polymer,7 inorganic material8 and magnetic nanoparticle9 has been well developed in recent years.10 Another effective strategy, known as one-phase catalysis and two-phase separation, provided an ideal and practical technique to combine the advantages of both homogeneous and heterogeneous catalysis, avoiding mass transfer limitation resulted from heterogeneous catalytic system.11 However, it is worthwhile to note that most of above-mentioned reactions, no matter whether in homogeneous or heterogeneous catalytic system, required cooperative catalysis of primary amine and various Brønsted acids with different acid strengths such as 2-nitrobenzoic acid or HFA (10–40 mol%) as a co-catalyst to achieve a satisfactory catalytic performance.12 Nevertheless, the simultaneous heterogenization of Brønsted acid and cinchona alkaloid-derived primary amine has been elusive owing to the lower catalytic performance compared to homogeneous counterparts in terms of activity, diastereoselectivity and enantioselectivity. Therefore, from the viewpoint of green chemistry, an efficient and recoverable supported organocatalyst possessing the multifunctionalities of enamine or iminium activation and acid catalysis is a compulsory task to meet the challenge of green chemistry.In the present study, based on solid acid catalytic property of phosphotungstic acid displayed especially in esterification,13 condensation,14 alkylation15 and alcoholysis,16 a novel type of phosphotungstic acid-supported multifunctional 9-amino(9-de-oxy)epi-cinchonidine organocatalyst possessing enamine activation and acid catalysis was developed by the facile direct precipitation of sodium tungstate with phosphonic acids with different arm lengths (n = 2, 4 and 6) instead of H3PO4 at various added P/W molar ratios from 0.5 to 4 (Scheme 1). As expected, in the case of the as-synthesized multifunctional organocatalysts CDNH2(n)–HPW, Brønsted acid and primary amine CDNH2(n) attached to the same backbone of phosphotungstic acid exhibited the concerted catalysis with high catalytic performances and good reusability in aqueous asymmetric aldol addition of different cyclic ketones to various aromatic aldehydes.
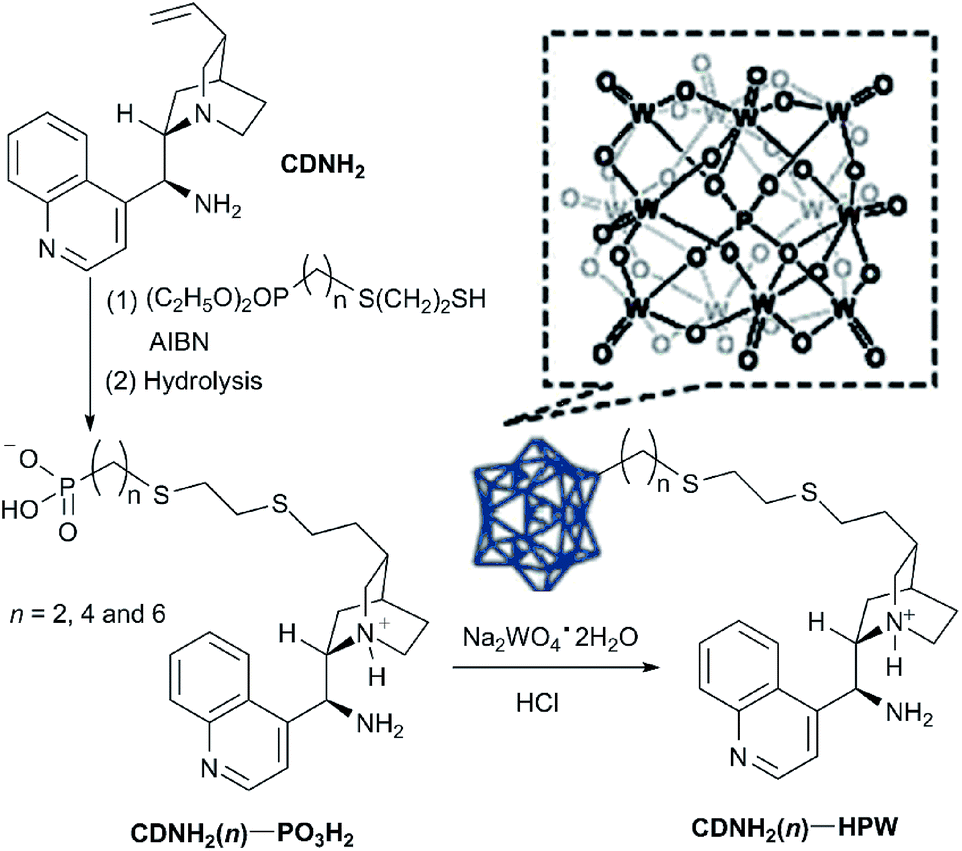 | ||
| Scheme 1 Synthesis of phosphotungstic acid-supported multifunctional CDNH2(n)–HPW possessing enamine activation and acid catalysis. | ||
Results and discussion
Preparation of phosphotungstic acid-supported CDNH2(n)–HPW
It was well known that 12-tungstophosphoric acid, H3PW12O40 (HPW), was readily prepared through the prolonged reflux of Na2WO4/H3PO4-mixed solution, acidification by HCl solution and then extraction with ethyl ether.17 After the replacement of organic CDNH2(n)–PO3H2 instead of inorganic H3PO4, CDNH2(n)–HPW/1, CDNH2(n)–HPW/2 and CDNH2(n)–HPW/4 could be directly precipitated out upon the added molar ratio of CDNH2(n)–PO3H2/Na2WO4 above 1. However, at the same procedure, no CDNH2(n4)–HPW/0.5 was directly precipitated out from reaction medium upon the added molar ratio of CDNH2(n4)–PO3H2/Na2WO4 below 1. To our delight, according to the synthetic procedure of 12-tungstophosphoric acid, CDNH2(n4)–HPW/0.5 could immediately precipitate out from solution after the medium pH was adjusted to 3–4 using HCl solution. In order to have a more comprehensive comparison of results, the effect of ageing the solids with added HCl in the preparation of CDNH2(n4)–HPW/1 and CDNH2(n4)–HPW/2 as examples was further investigated. Unfortunately, there was no marked difference in chemical composition and catalytic performance in aldol addition. Therefore, we did not set out the further investigation into the effect of HCl addition.Structure of phosphotungstic acid-supported CDNH2(n)–HPW
Unfortunately, our endeavors to cultivate single crystal to elucidate the real structure of CDNH2(n)–HPW were unsuccessful, possibly owing to the flexibility of attached bulky CDNH2(n)–PO3H2. Then, we did our endeavors to clarify the possible structure and true chemical composition of phosphotungstic acid-supported multifunctional CDNH2(n)–HPW by means of FT-IR, XRD, DTA, ICP and elemental analysis.It was well known that the IR spectra of neat H3PW12O40 had four characteristic IR bands approximately at 1080 (α-peak, P–O in the central tetrahedron), 980 (β-peak, terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 896 and 806 cm−1 (γ- and δ-peaks, asymmetric W–O–W) associated with pure Keggin structure18 and of M–O–P (M = metal). Furthermore, the free H–O–P stretching vibration in metal phosphonate was generally in the range of 1000–1100 cm−1.19 Although the characteristic absorption peaks of CDNH2(n)–HPW red-shifted about 50–80 cm−1, the hybrid CDNH2(n4)–HPW/1, CDNH2(n4)–HPW/2 and CDNH2(n4)–HPW/4 series had the similar four characteristic absorption peaks, respectively at 1031 and 983, 933 and 902, 832, 715 cm−1 (Fig. 1c–e). It was worthwhile to note that the vibrations of P–O in central tetrahedron and terminal WO bonds split into two bands respectively at 1031 and 983, 933 and 902 cm−1, possibly due to the formation of central P–O–W and terminal WO possessing diverse chemical environments upon the direct precipitation of Na2WO4 with CDNH2(n4)–PO3H2 at the added P/W molar ratios equal to or greater than 1. However, when the added molar ratio of P/W was 0.5, CDNH2(n4)–HPW/0.5 obtained in the pH = 3–4 range by means of hydrochloric acid showed the similar four characteristic peaks without split phenomenon at 1031, 933, 832, 715 cm−1 (Fig. 1b) as neat H3PW12O40, which implied that CDNH2(n4)–HPW/0.5 was more like the similar Keggin structure of H3PW12O40. Moreover, the C–H stretching vibrations of –CH2– and –CH– moieties in CDNH2 in the 2800–2980 cm−1 centered at 2930 cm−1 and characteristic phenyl ring in the 1620–1450 cm−1 range clearly corroborated the covalent attachment of CDNH2–PO3H2 to the hybrid backbone (see ESI†).
O), 896 and 806 cm−1 (γ- and δ-peaks, asymmetric W–O–W) associated with pure Keggin structure18 and of M–O–P (M = metal). Furthermore, the free H–O–P stretching vibration in metal phosphonate was generally in the range of 1000–1100 cm−1.19 Although the characteristic absorption peaks of CDNH2(n)–HPW red-shifted about 50–80 cm−1, the hybrid CDNH2(n4)–HPW/1, CDNH2(n4)–HPW/2 and CDNH2(n4)–HPW/4 series had the similar four characteristic absorption peaks, respectively at 1031 and 983, 933 and 902, 832, 715 cm−1 (Fig. 1c–e). It was worthwhile to note that the vibrations of P–O in central tetrahedron and terminal WO bonds split into two bands respectively at 1031 and 983, 933 and 902 cm−1, possibly due to the formation of central P–O–W and terminal WO possessing diverse chemical environments upon the direct precipitation of Na2WO4 with CDNH2(n4)–PO3H2 at the added P/W molar ratios equal to or greater than 1. However, when the added molar ratio of P/W was 0.5, CDNH2(n4)–HPW/0.5 obtained in the pH = 3–4 range by means of hydrochloric acid showed the similar four characteristic peaks without split phenomenon at 1031, 933, 832, 715 cm−1 (Fig. 1b) as neat H3PW12O40, which implied that CDNH2(n4)–HPW/0.5 was more like the similar Keggin structure of H3PW12O40. Moreover, the C–H stretching vibrations of –CH2– and –CH– moieties in CDNH2 in the 2800–2980 cm−1 centered at 2930 cm−1 and characteristic phenyl ring in the 1620–1450 cm−1 range clearly corroborated the covalent attachment of CDNH2–PO3H2 to the hybrid backbone (see ESI†).
 | ||
| Fig. 1 IR spectra of CDNH2(n4)–PO3H2 (a), CDNH2(n4)–HPW/0.5 (b), CDNH2(n4)–HPW/1 (c), CDNH2(n4)–HPW/2 (d) and CDNH2(n4)–HPW/4 (e). | ||
The X-ray powder diffraction patterns of neat H3PW12O40 displayed a set of well-resolved sharp diffraction peaks featured for the secondary crystal structure.18 However, all these characteristic sharp peaks disappeared and several broad diffraction peaks were observed in the case of CDNH2(n4)–HPW/2 and CDNH2(n4)–HPW/0.5 (Fig. 2), which indicated their non-crystal structure definitely different from the crystal Keggin structure of neat H3PW12O40. Compared with the characteristics of the Keggin unit with a sharp peak at about 6–10° for pristine H3PW12O40, both CDNH2(n4)–HPW/2 and CDNH2(n4)–HPW/0.5 presented the most intense and broad peak in the range of 2θ = 5–12°, stating clearly the existence of Keggin anions in the hybrids, in accordance with the above-mentioned IR spectra. Furthermore, the broad diffraction peaks in the 2θ = 15–40° and 45–70° ranges were also observed, suggesting that CDNH2(n4)–HPW/2 and CDNH2(n4)–HPW/0.5 existed in amorphous states without long-range order.20 This amorphous structure was in agreement with the HRTEM images for CDNH2(n4)–HPW/0.5 (Fig. 3). The TEM image showed the irregular particles of CDNH2(n4)–HPW/0.5 with the diameter of approximate 0.3–0.5 μm, whereas some irregular lattice fringes in different directions at the distances of 0.2–0.5 nm in the high resolution TEM image could be observed.
 | ||
| Fig. 2 Powder X-ray diffraction patterns of CDNH2(n4)–HPW/2 and CDNH2(n4)–HPW/0.5. | ||
 | ||
| Fig. 3 The TEM and HRTEM images of phosphotungstic acid-supported organo-catalyst CDNH2(n4)–HPW/0.5. | ||
Furthermore, no precipitate was obtained upon the direct mixing of sodium tungstate with 9-amino(9-deoxy)epi-cincho-nidine, which implied that the amino group in CDNH2(n) with-out phosphonic acid (–PO3H2) could not react sodium tungstate to afford the analogue of phosphotungstic acid. Combined the synthetic principle of phosphotungstic acid and the above-mentioned structural information obtained from FT-IR and XRD, the possible structure of the organic–inorganic hybrids CDNH2(n)–HPW was constructed by the replacement of the organic phosphonic acid in CDNH2(n)–PO3H2 instead of inorganic phosphate H3PO4 as the source of P to coordinate with a WO6− octahedra unit in parent phosphotungstic acid, and then the generated phosphotungstic acid was used as an inhert matrix to support organocatalyst CDNH2.
Chemical composition of phosphotungstic acid-supported CDNH2(n)–HPW
After 0.1000 g of CDNH2(n4)–HPW/0.5 was calcined for 4 h at 800 °C, the resulting solid was dissolved in 5.0 mL of HNO3 and 15 mL of DI water, pretreated via microwave digestion and then volumed to 100 mL. The content of wolfram in the mixture was detected by inductively coupled plasma atomic emission spectrometry (ICP-AES). The wolfram percent in CDNH2(n4)–HPW/0.5 was calculated to be 61.52%. In addition, the content of nitrogen (1.24%) in CDNH2(n4)–HPW/0.5, which was thrice the content of phosphorus in the same molecule, was detected by elemental analysis. Based on these results, the molar ratio of wolfram to phosphorus was calculated to be 11.2, which was very close to that of neat H3PW12O40. Mean-while, based to the nitrogen content, the loading capacity of CDNH2 in CDNH2(n4)–HPW/0.5 was also calculated to be 0.30 mmol g−1. According to the same method, the molar ratios of W/P and loading capacities of CDNH2 in CDNH2(n4)–HPW/1, CDNH2(n4)–HPW/2 and CDNH2(n4)–HPW/4 via direct precipitation were determined to be 10.0 and 0.33 mmol g−1, 9.8 and 0.34 mmol g−1, 10.4 and 0.32 mmol g−1, respectively. Further-more, with the decrease in the arm length (n) of CDNH2(n)–PO3H2 from 6 to 2, the W/P molar ratio in CDNH2(n2)–HPW/2 decreased to 8.8, and the loading capacity of CDNH2 increased to 0.38 mmol g−1. All the data was listed in Table 1. It was noticeable that the slightly lower W/P molar ratios (8.8–11.3) in CDNH2(n)–HPW than that of neat H3PW12O40 elucidated the possible coordination of CDNH2(n)–PO3H2 with MO6− octahedra in the synthetic process of these hybrids.| Cat. | Wa (wt%) | CDNH2(n)–PO3H2b (%/mmol g−1) | Hydration waterc,d (%/%) | Molar ratio (W/P)e |
|---|---|---|---|---|
| a Determined by ICP.b Determined by elemental analysis.c Determined automatic Karl Fischer moisture analyzer.d Determined by TGA below 120 °C.e Calculated according to the data of ICP and elemental analysis. | ||||
| CDNH2(n4)–HPW/0.5 | 61.52 | 15.5/0.30 | 2.9/3.8 | 11.2 |
| CDNH2(n4)–HPW/1 | 60.92 | 17.4/0.33 | 3.2/4.2 | 10.0 |
| CDNH2(n4)–HPW/2 | 60.98 | 17.7/0.34 | 3.1/4.4 | 9.8 |
| CDNH2(n4)–HPW/4 | 61.12 | 16.7/0.32 | 2.8/4.3 | 10.4 |
| CDNH2(n2)–HPW/2 | 61.03 | 18.6/0.38 | 3.0/5.3 | 8.8 |
| CDNH2(n6)–HPW/2 | 60.95 | 18.0/0.33 | 3.1/4.7 | 10.2 |
Thermal analysis was performed to monitor the decomposition profiles of CDNH2(n)–HPW. Using CDNH2(n4)–HPW/0.5 as an example, its TG, DTG and DSC curves were depicted in Fig. 4 and the others were shown in ESI.† The hydration water in CDNH2(n4)–HPW/0.5, which was verified by IR spectra with a broad and strong O–H stretching vibration in the 3200–3600 cm−1 range, was detected by automatic Karl Fischer moisture analyzer to be 2.9% of total weight. Compared with the data of weight loss (3.8%) by TG below 120 °C, it was found that the hydration water in CDNH2(n4)–HPW/0.5 obtained by Karl Fischer method had a relatively lower value with the 0.9% decrement, possibly owing to the effect of porous structure of back-bone. The protonized water and structural water lost in the 120–800 °C range,21 companied by the weight loss of organic moiety in CDNH2(n4)–PO3H2 with the exothermic peaks at 305, 432 and 600 °C due to the decomposition of CDNH2(n4)–HPW/0.5 and Keggin type anion. Based on the content of nitrogen (1.24%), the weight loss of organic moiety in CDNH2(n4)–HPW/0.5 was calculated to be 13.1%, and then the total weight loss of protonized water and structural water was summed to be 9.7%. On the whole, the chemical compositions of as-synthesized CDNH2(n)–HPW were approximate to each other (Table 1), although they had a big difference in the added P/W molar ratios from 0.5 to 4.0.
 | ||
| Fig. 4 The TG, DTG and DSC curves of supported multifunctional CDNH2(n4)–HPW/0.5. | ||
Porous structure of CDNH2(n)–HPW
The N2 adsorption–desorption isotherms of all samples, obtained at the temperature of 77 K, were shown in ESI.† Phosphotungstic acid-supported organocatalysts CDNH2(n4)–HPW/4, CDNH2(n4)–HPW/2, CDNH2(n2)–HPW/2 and CDNH2(n6)–HPW/2, prepared at the added P/W molar ratios equal to or greater than 2, exhibited the classic definition of a type III isotherm. Upon the decrease in the added P/W molar ratio below 2, CDNH2(n4)–HPW/1 and CDNH2(n4)–HPW/0.5 showed a step-height type VI isotherm, which was possibly related to the uniform porous structure.22 The BET-specific surface areas, average pore diameters and pore volumes of all samples were listed in Table 2. The overall trend along with the increased molar ratio of added P/W was towards the enhanced BET-specific surface areas, average pore diameters and pore volumes. Especially, when the added P/W molar ratios decreased from 4.0 to 0.5, the pore size distributions, obtained from BJH algorithm on adsorption isotherm, tended from the uneven micro mesoporous structures to the regular mesopores. From Fig. 5, it was found that CDNH2(n4)–HPW/0.5 and CDNH2(n4)–HPW/1 with the relatively low BET-specific surface areas, average pore diameters and pore volumes possessed the five regular meso-pores at 1.8, 2.9, 4.5, 6.8 and 15.0 nm, and the other adsorption Dv values were close to zero.| Cat. | Surface areab (m2 g−1) | Average pore diameterc (nm) | Pore volumec (×10−3 cm3 g−1) |
|---|---|---|---|
| a The samples were degassed at 105 °C for 5 h.b Based on the multipoint BET method.c Based on the desorption data using BJH method. | |||
| CDNH2(n4)–HPW/0.5 | 5.6 | 6.9 | 9.7 |
| CDNH2(n4)–HPW/1 | 13.8 | 8.4 | 8.0 |
| CDNH2(n4)–HPW/2 | 18.2 | 23.8 | 108.2 |
| CDNH2(n4)–HPW/4 | 22.1 | 15.2 | 84.2 |
| CDNH2(n2)–HPW/2 | 23.5 | 26.8 | 157.3 |
| CDNH2(n6)–HPW/2 | 16.5 | 68.7 | 42.8 |
 | ||
| Fig. 5 The pore size distributions (PSDs) of CDNH2(n2)–HPW/2 (a), CDNH2(n6)–HPW/2 (b), CDNH2(n4)–HPW/4 (c), CDNH2(n4)–HPW/2 (d), CDNH2(n4)–HPW/1 (e) and CDNH2(n4)–HPW/0.5 (f). | ||
Acid capacity and acid strength
Generally, NH3–TPD was employed to reveal acidic site nature of solid acid.23 Unfortunately, owing to the disturbance of weight loss resulted from the organic moieties in CDNH2(n)–HPW in the high 200–600 °C temperature range, the acidic site nature was incapable of being investigated by terms of NH3–TPD technique. Herein, the acid capacities of CDNH2(n)–HPW were determined by simple and convenient acid–base titration, and the acid–base titration curves by NaOH aqueous solution (0.05 mol L−1) were presented in Fig. 6 and Table S1 (ESI†). It was found that synthetic method had a great influence on the acid capacities of CDNH2(n)–HPW. Compared with CDNH2(n4)–HPW/4 (45.0 mmol g−1), CDNH2(n4)–HPW/2 (40.5 mmol g−1) and CDNH2(n4)–HPW/1 (12.0 mmol g−1) prepared via direct precipitation at the added P/W molar ratio equal to or greater than 1, the highest acid capacity (61.5 mmol g−1) was observed for CDNH2(n4)–HPW/0.5 obtained at the lowest added P/W molar ratio in the pH = 3–4 ranges assisted by HCl, although CDNH2(n4)–HPW/0.5 possessed the lowest surface area, average pore diameter and pore volume. Moreover, the arm lengths (n) of attached CDNH2(n)–PO3H2 had no impact on the acid capacity. It was observed that CDNH2(n2)–HPW/2 (39.5 mmol g−1), CDNH2(n4)–HPW/2 (40.5 mmol g−1) and CDNH2(n6)–HPW/2 (39.0 mmol g−1) exhibited the identical level of acid capacity. | ||
| Fig. 6 The acid–base titration curves of CDNH2(n2)–HPW/2 (a), CDNH2(n6)–HPW/2 (b), CDNH2(n4)–HPW/0.5 (c), CDNH2(n4)–HPW/1 (d), CDNH2(n4)–HPW/2 (e) and CDNH2(n4)–HPW/4 (f) by aqueous NaOH solution (0.05 mol L−1). | ||
The acid strengths of phosphotungstic acid-supported organocatalysts CDNH2(n)–HPW were examined by Hammett indicator method (Table S2, ESI†). Probably owing to the more like Keggin structure of CDNH2(n4)–HPW/0.5 verified by the IR spectra (Fig. 1), it was found that CDNH2(n4)–HPW/0.5 possessed the strongest acid strength (H0 ≤ 0.8). Among the as-synthesized multifunctional organocatalysts CDNH2(n)–HPW, CDNH2(n4)–HPW/1 had the weakest acid strength (H0 ≤ 3.86). Unfortunately, only based on the structural information obtained from IR and XRD, the reason can hardly be unconvincingly explained.
Asymmetric aldol addition
To evaluate the multifunctional catalytic performance and reusability of CDNH2(n)–HPW simultaneously containing attached organocatalyst CDNH2(n) and Brøsted acid, the asymmetric aldol addition of cyclohexanone to benzaldehydes was selected as a model reaction at 15 °C. Generally, aldol reaction, catalyzed by 9-amino(9-deoxy)epi-cinchona alkaloid, needs an additional acid co-catalyst such as F3CSO3H to achieve a desired excellent catalytic performance. Excitedly, owing to the strong acidity of CDNH2(n)–HPW resulted from the backbone of phosphotungstic acid verified by Hammett indicator, it was highlighted that the aldol reaction smoothly carried out without the addition of an external acidic additive.From Table 3, it was found that the arm length (n) and acid capacity of CDNH2(n)–HPW had a significant effect on the co-operative catalysis of 9-amino(9-deoxy)epi-cinchonidine and Brøsted acid. The optimal arm length (n) was n = 4 in terms of the excellent yield (95%) and good stereoselectivity (anti/syn = 86/14, 94% ee anti, entries 3, 5 and 6). Notably, when the arm length increased to n = 6, the cooperative catalysis between 9-amino(9-de-oxy)epi-cinchonidine and Brøsted acid was getting weaker and resulted in the unsatisfactory catalytic performances (81% yield, anti/syn = 86/14, 91% ee anti, entry 6). Further-more, owing to the highest acid capacity (61.5 mmol g−1) and regular mesopore structure (Fig. 5), CDNH2(n4)–HPW/0.5 afforded the excellent stereoselectivity (anti/syn = 93/7, 96% ee anti, entry 1) in 96% yield. Unfortunately, along with the decrease of acid capacity, the more low yields and stereoselectivities were observed for CDNH2(n4)–HPW prepared via direct precipitation at the added P/W molar ratio equal to or greater than 1 (entries 2–4).
| Entry | Cat. | Yieldb (%) | Dr (anti/syn)c | % ee (anti)d |
|---|---|---|---|---|
| a Reaction conditions: cat. (40 mg, 0.012–0.014 mmol), cyclohexanone (0.5 mL, 4.8 g, 49.0 mmol), p-nitrobenzaldehyde (30.2 mg, 0.2 mmol), water (0.5 mL), 15 °C, 72 h.b Isolated yield.c Determined by 1H NMR.d Determined by chiral HPLC. | ||||
| 1 | CDNH2(n4)–HPW/0.5 | 96 | 93/7 | 96 |
| 2 | CDNH2(n4)–HPW/1 | 93 | 89/11 | 95 |
| 3 | CDNH2(n4)–HPW/2 | 95 | 86/14 | 94 |
| 4 | CDNH2(n4)–HPW/4 | 93 | 86/14 | 94 |
| 5 | CDNH2(n2)–HPW/2 | 90 | 85/15 | 92 |
| 6 | CDNH2(n6)–HPW/2 | 81 | 84/16 | 91 |
Using CDNH2(n4)–HPW/0.5 with the best catalytic performance as a example, the optimized protocol was expanded to aldol reaction of cyclohexanone and hydroxyacetone to a wide variety of aldehydes. From Table 4, o, m and p-substituted benzaldehydes bearing electron-withdrawing substituents (–X and –NO2) afforded the good yields (71–96%) and excellent stereoselectivities (anti/syn = 98–94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2–6, 97–87% ee, entries 1–3 and 7–9). For m and p-CN substituted benzaldehydes, the excellent enantioselectivities and moderate diasteroselectivities were achieved in good yields (entries 5 and 6). For some un-fathomable reason, no aldol adduct was obtained in the aldol addition of cyclohexanone to o-CN substituted benzaldehyde (entry 4). Unfortunately, the aldol reaction is dramatically dependent on the electronic effect of the substituent at the aromatic ring like its homogeneous catalysis. The substrates with electron-donating o-, m- and p-CH3 and OCH3 groups at the aromatic ring gave the less unsatisfactory yields (0–59%, entries 10–15), which was attributed to not enough acid strength and capacity for CDNH2(n4)–HPW/0.5 to favorably catalyze the aldol addition. Furaldehyde was also employed in the aldol reaction which showed the high yield (92%) and enantioselectivity far from satisfactory (69% ee) (entry 16). Furthermore, we tested the aldol reaction of hydroxyacetone with benzaldehydes bearing electron-withdrawing o, m or p-nitro substituent and 2,4-dinitro multiple substituents. All of them afforded the high yields (89–95%), but unfortunately, only the moderate syn/anti = 72–81/19–28 and moderate to good 77–91% ee obtained for benzaldehydes bearing o, m or p-nitro substituent. To our delight, the excellent stereoselectivities (syn/anti = 94/6 and 95% ee) were achieved for benzaldehyde bearing 2,4-dinitro substituents.
:2–6, 97–87% ee, entries 1–3 and 7–9). For m and p-CN substituted benzaldehydes, the excellent enantioselectivities and moderate diasteroselectivities were achieved in good yields (entries 5 and 6). For some un-fathomable reason, no aldol adduct was obtained in the aldol addition of cyclohexanone to o-CN substituted benzaldehyde (entry 4). Unfortunately, the aldol reaction is dramatically dependent on the electronic effect of the substituent at the aromatic ring like its homogeneous catalysis. The substrates with electron-donating o-, m- and p-CH3 and OCH3 groups at the aromatic ring gave the less unsatisfactory yields (0–59%, entries 10–15), which was attributed to not enough acid strength and capacity for CDNH2(n4)–HPW/0.5 to favorably catalyze the aldol addition. Furaldehyde was also employed in the aldol reaction which showed the high yield (92%) and enantioselectivity far from satisfactory (69% ee) (entry 16). Furthermore, we tested the aldol reaction of hydroxyacetone with benzaldehydes bearing electron-withdrawing o, m or p-nitro substituent and 2,4-dinitro multiple substituents. All of them afforded the high yields (89–95%), but unfortunately, only the moderate syn/anti = 72–81/19–28 and moderate to good 77–91% ee obtained for benzaldehydes bearing o, m or p-nitro substituent. To our delight, the excellent stereoselectivities (syn/anti = 94/6 and 95% ee) were achieved for benzaldehyde bearing 2,4-dinitro substituents.
| Entry | Main product | Yieldb (%) | Drc (anti/syn) | % eed |
|---|---|---|---|---|
| a Reaction conditions: CDNH2(n4)–HPW/0.5 (40 mg, 0.012 mmol, 6.0 mol%), cyclohexanone (0.5 mL, 4.8 g, 49.0 mmol), various benzaldehyde derivatives (0.2 mmol), water (0.5 mL), 15 °C, 72 h.b Isolated yield.c Determined by 1H NMR.d Determined by chiral HPLC. | ||||
| 1 | 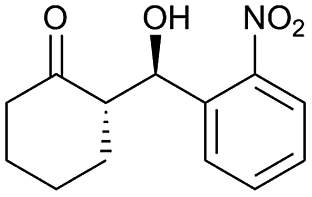 |
83 | 98/2 | 95 |
| 2 | 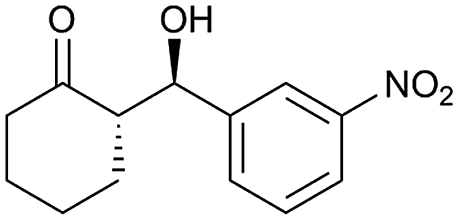 |
90 | 93/7 | 96 |
| 3 | 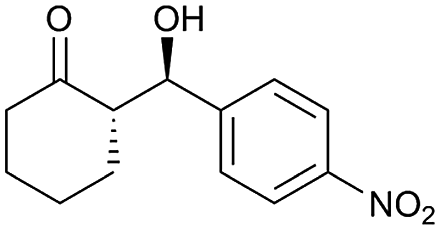 |
96 | 93/7 | 96 |
| 4 |  |
— | — | — |
| 5 |  |
80 | 84/16 | 95 |
| 6 | 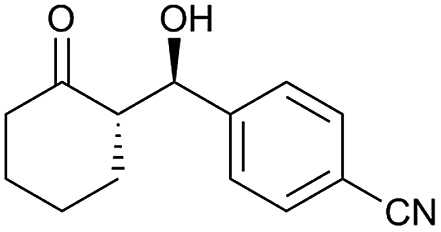 |
76 | 85/15 | 98 |
| 7 |  |
74 | 98/2 | 97 |
| 8 | 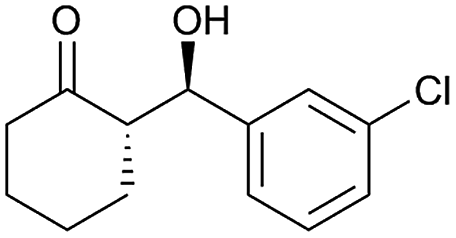 |
71 | 95/5 | 94 |
| 9 | 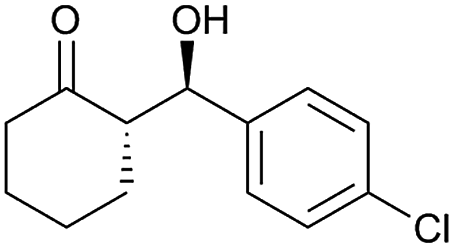 |
75 | 94/6 | 87 |
| 10 |  |
35 | 85/15 | 97 |
| 11 |  |
59 | 82/18 | 95 |
| 12 |  |
20 | 91/9 | 88 |
| 13 | 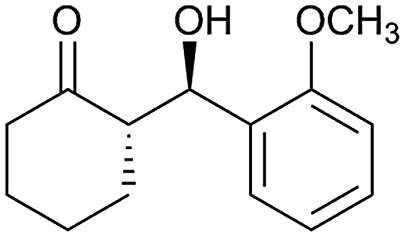 |
19 | 78/22 | 90 |
| 14 | 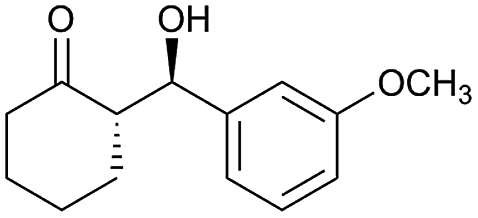 |
36 | 86/14 | 76 |
| 15 |  |
— | — | — |
| 16 |  |
92 | 86/14 | 69 |
| 17 |  |
95 | 19/81 | 88 |
| 18 |  |
90 | 30/70 | 77 |
| 19 |  |
89 | 28/72 | 91 |
| 20 |  |
94 | 6/94 | 95 |
On the whole, compared to the ever-reported heterogeneous catalytic aldol additions, CDNH2(n4)–HPW/0.5 without the assistance of additional Brøsted acid afforded the higher stereoselectivity than the other 9-amino(9-deoxy)epi-cinchona alkaloid supported on inorganic backbones such as zirconium phosphonate8a and Fe3O4 MNPs,9 especially in the aldol addition of cyclohexanone to o, m and p-substituted benzaldehydes bearing electron-withdrawing (–X, –NO2 and –CN) substituents and hydroxyacetone to 2,4-dinitrobenzaldehyde.
Reusability of phosphotungstic acid-supported CDNH2(n4)–HPW/0.5
After the completion of aldol addition of cyclohexanone to o-nitrobenzaldehyde, the reaction mixture was centrifuged, decanted, and the recovered catalyst CDNH2(n4)–HPW/0.5 was reused directly for the next run after being washed with ethyl acetate (2.0 mL × 3). To our delight, it was found that the constant excellent stereoselectivity (96% ee and anti/syn = 93/7) and yield (96%) were maintained in five consecutive cycles. Even at the eighth run, there was only a slight drop in the yield (96% → 94%), diastereoselectivity (anti/syn = 93/7 → 89/11) and enantioselectivity (96% ee → 94%). After the completion of aldol reaction, the aqueous phase was evaporated, pretreated via microwave digestion in HNO3 (1.0 mL) and DI water (3.0 mL) and volumed to 10 mL. The content of wolfram in the resulting solution was checked by ICP-AES. It was found that there was no leaching of wolfram during aldol reaction. However, after being reused seven times, the sulphur content in CDNH2(n4)–HPW/0.5 was found to decrease from 1.91% to 1.85% detected by elemental analysis, which elucidated that 3.1% weight loss of CDNH2(n4) resulted in the decrease of CDNH2(n4)–HPW/0.5 in catalytic performances.Conclusions
In summary, we had developed a novel type of supported multifunctional organocatalysts containing 9-amino(9-deoxy)epi-cinchonidine and Brønsted acid on the backbone of phosphotungstic acid via facile precipitation of sodium tungstate with organophosphonic acid for the first time, and successfully applied these supported organocatalysts in the asymmetric aldol addition of cyclohexanone and hydroxyacetone to various aromatic aldehydes bearing electron-withdrawing substituents with good to excellent catalytic performances. From the view-point of green chemistry, these phosphotungstic acid-supported organocatalysts achieved the simultaneous green circulation of organocatalyst and Brønsted acid and possessed a good reusability with no significant loss of catalytic performances in the five consecutive asymmetric aldol addition. Furthermore, the authentic structure of CDNH2(n)–HPW and their applications for other challenging enantioselective cooperative catalysis is under the further investigation.Experimental
Chemicals
All the chemicals including hydrochloric acid (36.5–38.0%) and sodium tungstate dehydrate (Na2WO4·2H2O, ≥99.0%) were purchased and used without any further purification. Phosphonic acids CDNH2(n)–PO3H2 (n = 2, 4 and 6) were synthesized by the click reaction of 9-amino(9-deoxy)epi-cinchonidine24 with HS(CH2)2S(CH2)nPO(OC2H5)2 and subsequent hydrolysis of ethyl phoshonate according to the ref. 8a and 9.Characterization
1H and 13C NMR spectra were conducted using a Bruker av-600 NMR instrument, in which all chemical shifts were reported down-field in ppm relative to the hydrogen and carbon resonances of TMS. FT-IR spectroscopy was performed on a Perkin-Elmer model GX spectrometer using KBr pellet. Thermogravimetry-differential thermal analysis was recorded on SBTQ600 thermal analyzer at a heating rate of 10 °C min−1 from 40 °C to 800 °C using N2 as a protective gas (100 mL min−1). Elemental analysis was obtained using a vario Micro cube elemental analyzer. The morphology was observed by Tecnai G2 F20 transmission electron microscope, operated at 200 kV. X-ray powder diffraction patterns were detected on an XRD-7000 S/L instrument: Cu-Kα radiation, X-ray tube settings of 40 kV/30 mA and a step size of 2° min−1 in the 2θ range of 5–70°. N2 adsorption–desorption isotherm was carried out at 77.4 K using an Autosorb-1 apparatus (Quantachrome), in which the sample was degassed at 120 °C for 12 h before measurement and specific surface area and pore diameter were calculated by a BET method and BJH model, respectively. The water content in sample was detected by means of MA-1 automatic Karl Fischer moisture analyzer. The anti/syn ratios were determined by peak area ratios of syn- and anti-CHOH protons, respectively at δ 5.3–5.9 ppm and δ 4.7–5.4 ppm in 1H NMR (see ESI†), and the enantiomeric excess (% ee) was determined by Agilent LC-1200 HPLC with a 220 nm or 254 nm UV-vis detector using Daicel Chiralpak chiral OD-H column (4.6 mm × 25 cm), eluting with n-hexane/isopropanol (95/5) at a flow rate of 0.5 mL min−1 under 20 °C.Preparation of various supported organocatalysts CDNH2(n)–HPW
CDNH2(n4)–PO3H2 (n = 4) (30.5 mg, 0.05 mmol) in DI water (0.5 mL) was added 0.5 mL of aqueous sodium tungstate solution (29.4 mg, 0.1 mmol), and mixed under stirring at room temperature to afford a transparent solution. The medium pH was ad-justed to 3–4 using hydrochloric acid (36.5–38.0%), and light yellow solid precipitated immediately. After being aged for another 6 h, the resulting precipitate was filtered, washed with DI water (2 mL × 3) and dried under vacuum at 50 °C to afford phosphotungstic acid-supported multifunctional organocatalyst, donated as CDNH2(n4)–HPW/0.5 (48 mg, 81%). Found C 8.86, H 1.23, N 1.24, S 1.91 by elemental analysis and W 61.52 by ICP-AES for CDNH2(n4)–HPW/0.5.According to the same above-mentioned procedure, when the added molar ratios of CDNH2(n4)–PO3H2 to sodium tungstate equalled to 1, 2 and 4, corresponding supported multifunctional organocatalysts CDNH2(n4)–HPW/1 (27 mg, 45%), CDNH2(n4)–HPW/2 (30 mg, 50%) and CDNH2(n4)–HPW/4 (34 mg, 57%) could precipitated out directly in the absence of hydrochloric acid (36.5–38.0%). Found C 10.01, H 1.26, N 1.40, S 2.14 by elemental analysis and W 60.92 by ICP-AES for CDNH2(n4)–HPW/1; found C 10.14, H 1.30, N 1.42, S 2.18 by elemental analysis and W 60.98 by ICP-AES for CDNH2(n4)–HPW/2, and found C 9.58, H 1.27, N 1.34, S 2.06 by elemental analysis and W 61.12 by ICP-AES for CDNH2(n4)–HPW/4.
Furthermore, CDNH2(n)–PO3H2 with the different arm lengths (n = 2 and 6) were used instead of CDNH2(n4)–PO3H2 (n = 4) to afford CDNH2(n2)–HPW/2 (28 mg, 47%) and CDNH2(n6)–HPW/2 (39 mg, 65%) according to the same procedure as CDNH2(n4)–HPW/2. Found C 12.20, H 1.52, N 1.58, S 2.43 by elemental analysis and W 61.03 by ICP-AES for CDNH2(n2)–HPW/2, and found C 10.57, H 1.39, N 1.37, S 2.11 by elemental analysis and W 60.95 by ICP-AES for CDNH2(n6)–HPW/2.
Determination of acid capacity
In 100 mL round-bottom flask, the sample (100 mg) was immersed in 10 wt% aqueous sodium chloride (50 mL), well-dispersed under sonic radiation for 30 min, and the medium pH was monitored by a pH meter till pH was constant (about 24 h). The acid content in the resulting solution was determined by acid–base titration method using NaOH solution (0.05 mol L−1).General procedure for asymmetric aldol reaction
In a 25 mL microreactor, CDNH2(n4)–HPW/0.5 (40 mg, 0.012 mmol of CDNH2(n4)), DI water (0.5 mL) and cyclohexanone (0.5 mL) were well mixed at 15 °C for about 15 min. Subsequently, p-nitrobenzaldehyde (30.2 mg, 0.2 mmol) was added, and the resulting mixture was allowed to react at 15 °C for 48–72 h. The reaction process was monitored by TLC. After the completion of the reaction, CDNH2(n4)–HPW/0.5 was recovered by centrifugal separation, and the centrifugate was extracted with ethyl acetate (4 mL × 3). The combined organic phases were dried over anhydrous Na2SO4 and evaporated under reduced pressure to afford a crude product, which was purified by flash column chromatography and eluted with petroleum ether/ethyl acetate (v/v = 10/1 → 2/1) to give the pure 2-(hydroxy(4-methoxyphenyl)methyl)cyclohexanone.Acknowledgements
We are grateful to the Fundamental Research Funds for the Central Universities of China (XDJK2015D028).Notes and references
- (a) B. L. Zheng, Q. Z. Liu, C. S. Guo, X. L. Wang and L. He, Org. Biomol. Chem., 2007, 5, 2913 RSC; (b) Q. Guo and J. C. G. Zhao, Tetrahedron Lett., 2012, 53, 1768 CrossRef CAS; (c) J. Zhou, V. Wakchaure, P. Kraft and B. List, Angew. Chem., Int. Ed., 2008, 47, 7656 CrossRef CAS PubMed; (d) A. Kumar and S. S. Chimni, Eur. J. Org. Chem., 2013, 4780 CrossRef CAS; (e) Y. H. Lam and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 2116 CrossRef CAS PubMed; (f) P. Li, J. Zhao, F. Li, A. S. C. Chan and F. Y. Kwong, Org. Lett., 2010, 12, 5616 CrossRef CAS PubMed.
- (a) G. K. Prakash, F. Wang, Z. Zhang, C. F. Ni, R. Haiges and G. A. Olah, Org. Lett., 2012, 14, 3260 CrossRef CAS PubMed; (b) W. J. Liu, D. S. Mei, W. Wang and W. H. Duan, Tetrahedron Lett., 2013, 54, 3791 CrossRef CAS; (c) A. Lee, C. M. Reisinger and B. List, Adv. Synth. Catal., 2012, 354, 1701 CrossRef CAS; (d) X. Tian, Y. K. Liu and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 6439 CrossRef CAS PubMed; (e) L. Liu, D. Y. Wu, X. M. Li, S. N. Wang and W. Wang, Chem. Commun., 2012, 48, 1692 RSC; (f) W. Yang, K. Z. Jiang, X. Lu, H. M. Yang, L. Li, Y. X. Lu and L. W. Xu, Chem.–Asian J., 2013, 8, 1182 CrossRef CAS PubMed; (g) J. Christensen, Ł. Albrecht and K. A. Jørgensen, Chem.–Asian J., 2013, 8, 648S CrossRef PubMed.
- (a) M. S. Jadhav, P. Righi, E. Marcantoni and G. Bencivenni, J. Org. Chem., 2012, 77, 2667 CrossRef CAS PubMed; (b) N. Demoulin, O. Lifchits and B. List, Tetrahedron, 2012, 68, 7568 CrossRef CAS; (c) O. Lifchits, N. Demoulin and B. List, Angew. Chem., Int. Ed., 2011, 50, 9680 CrossRef CAS PubMed.
- (a) A. Gualandi, D. Petruzziello, E. Emer and P. G. Cozzi, Chem. Commun., 2012, 48, 3614 RSC; (b) M. G. Capdevila, E. Emer, A. Gualandi, D. Petruzziello, S. Grilli and P. G. Cozzi, ChemCatChem, 2012, 4, 968 CrossRef CAS.
- (a) X. Feng, Z. Zhou, R. Zhou, Q. Q. Zhou, L. Dong and Y. C. Chen, J. Am. Chem. Soc., 2012, 134, 19942 CrossRef CAS PubMed; (b) J. Y. Liang, Q. Chen, L. P. Liu, X. X. Jiang and R. Wang, Org. Biomol. Chem., 2013, 11, 1441 RSC.
- (a) J. D. Duan and P. F. Li, Catal. Sci. Technol., 2014, 4, 311 RSC; (b) C. M. R. Volla, L. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390 CrossRef CAS PubMed.
- (a) R. Porta, M. Benaglia, F. Coccia, F. Cozzi and A. Puglisi, Adv. Synth. Catal., 2015, 357, 377 CrossRef CAS; (b) R. Porta, F. Coccia, R. Annunziata and A. Puglisi, ChemCatChem, 2015, 7, 1490 CrossRef CAS; (c) J. Q. Zhou, J. W. Wan, X. B. Ma and W. Wang, Org. Biomol. Chem., 2012, 10, 4179 RSC; (d) J. Izquierdo, C. Ayats, A. H. Henseler and M. A. Pericàs, Org. Biomol. Chem., 2015, 13, 4204 RSC.
- (a) W. Wang, X. B. Ma, J. W. Wan, J. Cao and Q. Tang, Dalton Trans., 2012, 41, 5715 RSC; (b) P. García-García, A. Zagdoun, C. Copéret, A. Lesage, U. Díıaza and A. Corma, Chem. Sci., 2013, 4, 2006 RSC.
- J. W. Wan, L. ding, T. Wu, X. B. Ma and Q. Tang, RSC Adv., 2014, 4, 38323 RSC.
- M. Bartók, Catal. Rev.: Sci. Eng., 2015, 57, 192 Search PubMed.
- J. W. Wan, Z. W. Zhao, F. L. Wang and X. B. Ma, Eur. J. Org. Chem., 2015, 26, 5755 CrossRef.
- (a) Y. H. Lam and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 9556 CrossRef CAS PubMed; (b) J. D. Duan and P. F. Li, Catal. Sci. Technol., 2014, 4, 311 RSC; (c) Y. Q. Wang, X. Y. Cui, Y. Y. Rena and Y. N. Zhang, Org. Biomol. Chem., 2014, 12, 9101 RSC; (d) H. Zhao, Y. B. Lan, Z. M. Liu, Y. Wang, X. W. Wang and J. C. Tao, Eur. J. Org. Chem., 2012, 1935 CrossRef CAS; (e) P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 9748 CrossRef CAS PubMed.
- (a) Zillillah, T. A. Ngu and Z. Li, Green Chem., 2014, 16, 1202 RSC; (b) S. H. Zhu, X. Q. Gao, F. Dong, Y. L. Zhu, H. Y. Zheng and Y. W. Li, J. Catal., 2013, 306, 155 CrossRef CAS; (c) W. H. Zhang, Y. Leng, D. R. Zhu, Y. J. Wu and J. Wang, Catal. Commun., 2009, 11, 151 CrossRef CAS.
- (a) Q. Deng, G. K. Nie, L. Pan, J. J. Zou, X. W. Zhang and L. Wang, Green Chem., 2015, 17, 4473 RSC; (b) M. Saikia, D. Bhuyan and L. Saikia, Appl. Catal., A, 2015, 505, 501 CrossRef CAS; (c) F. Fache, F. Cros, O. Piva and F. Lefebvre, Synth. Commun., 2012, 42, 2098 CrossRef CAS.
- (a) Z. K. Zhao and X. H. Wang, Appl. Catal., A, 2015, 503, 103 CrossRef CAS; (b) X. L. Sheng, Y. M. Zhou, Y. W. Zhang, Y. Z. Duan and M. W. Xue, Catal. Lett., 2012, 142, 360 CrossRef CAS; (c) K. Shimizu, K. Niimi and A. Satsuma, Appl. Catal., A, 2008, 349, 1 CrossRef CAS.
- L. H. Wee, F. Bonino, C. Lamberti, S. Bordiga and J. A. Martens, Green Chem., 2014, 16, 1351 RSC.
- (a) G. A. Tsigdinos and C. J. Hallada, Inorg. Chem., 1968, 7, 437 CrossRef CAS; (b) D. R. Park, S. H. Song, U. G. Hong, J. G. Seo, J. C. Jung and I. K. Song, Catal. Lett., 2009, 132, 363 CrossRef CAS; (c) D. R. Park, S. Park, Y. Bang and I. K. Song, Appl. Catal., A, 2010, 373, 201 CrossRef CAS.
- (a) P. Staiti, S. Freni and S. Hocevar, J. Power Sources, 1999, 79, 250 CrossRef CAS; (b) X. L. Sheng, J. Kong, Y. M. Zhou, Y. W. Zhang, Z. W. Zhang and S. J. Zhou, Microporous Mesoporous Mater., 2014, 187, 7 CrossRef CAS; (c) Y. Leng, P. P. Zhao, M. J. Zhang and J. Wang, J. Mol. Catal. A: Chem., 2012, 358, 67 CrossRef CAS; (d) W. X. Gao, X. Y. Sun, H. L. Niu, X. J. Song, K. G. Li, H. C. Gao, W. X. Zhang, J. H. Yu and M. J. Jia, Microporous Mesoporous Mater., 2015, 213, 59 CrossRef CAS.
- J. C. Amicangelo and W. R. Leenstra, Inorg. Chem., 2005, 44, 2067 CrossRef CAS PubMed.
- M. Nagai, K. Kobayashi and Y. Nakajima, Solid State Ionics, 2000, 136, 249 CrossRef.
- X. Tong, N. Q. Tian, W. Wu, W. M. Zhu, Q. Y. Wu, F. H. Cao, W. F. Yan and A. B. Yaroslavtsev, J. Phys. Chem. C, 2013, 117, 3258 CAS.
- K. S. W. Sing, G. D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemiewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- (a) Z. K. Zhao and X. H. Wang, Appl. Catal., A, 2015, 503, 103 CrossRef CAS; (b) R. Liu, X. Z. Niu, X. N. Xia, Z. B. Zeng, G. Z. Zhang and Y. B. Lu, RSC Adv., 2015, 5, 62394 RSC; (c) B. Viswanadham, V. Pavankumar and K. V. R. Chary, Catal. Lett., 2014, 144, 744 CrossRef CAS; (d) T. Wu, J. W. Wan and X. B. Ma, Chin. J. Catal., 2015, 36, 425 CrossRef CAS.
- (a) H. Brunner, J. Biigler and B. Nuber, Tetrahedron: Asymmetry, 1995, 6, 1699 CrossRef CAS; (b) H. L. Cui and F. Tanaka, Chem.–Eur. J., 2013, 19, 6213 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TGA, TEM and N2 adsorption–desorption isotherm of catalysts, 1H, 13C NMR and HPLC spectra of intermediates and products. See DOI: 10.1039/c6ra08909g |
| This journal is © The Royal Society of Chemistry 2016 |