
A quantum chemical approach towards understanding stability and tautomerism of 2-imino-2H-pyran derivatives†

Abstract
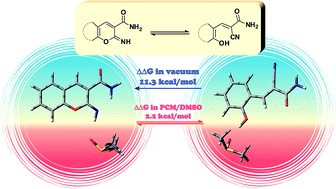
The ring-chain tautomerism of 2-imino-2H-pyran derivatives annelated with an aromatic or aliphatic ring and their transformation into corresponding 2-pyridons were theoretically studied based on the relative stabilities of two series of model isomers calculated by the DFT B3LYP/aug-cc-pVDZ method. This approach, augmented by the analysis of specific interactions between the solute and solvent molecules (DMSO or acetone) in 1 : 1 complexes and simulation of the bulk solvent medium by PCM, allowed a good agreement with the experimental data for 2-imino-2H-coumarines and explained their ring-chain tautomerism in different media. The main reason governing the stabilization of the open-chain isomers was found to be the specific intermolecular interaction of the hydroxyl groups of these isomers with a solvent molecule.
 Please wait while we load your content...
Please wait while we load your content...

