
Facile synthesis of 3-amino-5-aryl-1,2,4-oxadiazoles via PIDA-mediated intramolecular oxidative cyclization†
Abstract
A mild and efficient method for the synthesis of 3-amino-5-aryl-1,2,4-oxadiazole by intramolecular cyclization using PhI(OAc)2 (PIDA) as an oxidant is developed. Various 3-amino-5-aryl-1,2,4-oxadiazoles are prepared in moderate to good yields, and the PIDA-mediated N–O bond formation mechanism is suggested. In view of the readily available starting materials, operational simplicity, high functionality tolerance, and low toxicity, this protocol provides a novel synthetic strategy for 1,2,4-oxadiazoles.
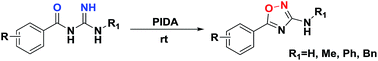
 Please wait while we load your content...
Please wait while we load your content...

