
Trapping oxygen in hierarchically porous carbon nano-nets: graphitic nitrogen dopants boost the electrocatalytic activity†
Li-Na Han,
Xiao Wei,
Bing Zhang,
Xin-Hao Li*,
Qian-Cheng Zhu,
Kai-Xue Wang and
Jie-Sheng Chen*
School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: xinhaoli@sjtu.edu.cn; chemcj@sjtu.edu.cn
First published on 1st June 2016
Abstract
Rational control in both the chemical structures of nitrogen dopants and the mechanical structure in the mesoscale is the key point for optimizing the electrocatalytic activities of carbon nanocatalysts. Here we report the fabrication of N-doped carbon nano-nets (denoted g-N-MM-Cnet) with precise dopant distribution at graphitic sites and multiple-scale pore dimensions via supramolecular assemblies of block copolymer P123. The enriched graphitic nitrogen dopants, high surface area (1947 m2 g−1) and microporous–mesoporous bimodal nanopores in g-N-MM-Cnet make it an excellent bifunctional electrocatalyst for oxygen evolution and reduction reactions to construct rechargeable and ultra-stable (cycling lifetime: 491 h) two-electrode alkaline zinc–air batteries with a high energy density of up to 866.4 W h kgZn−1 at 5 mA cm−2.
1 Introduction
Nitrogen-doped nanocarbons have received much attention due to their great potential as alternative catalysts to Pt/C for the oxygen reduction reaction (ORR)1,2 and oxygen evolution reaction (OER).3,4 Both the ORR and OER are of paramount importance for large-scale commercialization of fuel cells,5 metal–air secondary batteries6,7 and even chlor-alkali processes.8 Compared with bench-marked noble metal electrocatalysts, metal-free N-doped carbons are preferred due to their good durability, and most importantly, low cost,9–14 though metal-free electrocatalysts still suffer from low OER activity at the moment. It is difficult to get acceptable OER and ORR activities and stabilities together within the same carbon material, even in metal-containing catalysts. As an alternative, OER catalysts and ORR catalysts are usually loaded separately at two electrodes to achieve stable oxygen evolution and reduction processes for constructing rechargeable three-electrode metal–air batteries, resulting in a much higher cost but lower energy intensity of the complete batteries.6,7,14–17In order to elevate the activity and stability of N-doped carbon electrocatalysts for cheap, rechargeable and durable two-electrode metal–air batteries, the chemical structures of nitrogen heteroatoms and pore dimensions should be rationally designed to simultaneously optimize the activity and mass/charge transfer efficiency of nanocarbons for both ORR and OER.18–21 There have been a number of long-term studies on controlling both the content and structure of nitrogen dopants in carbon based nanomaterials to boost their catalytic activity, not limited to ORR and OER performance.22–25 For the nitrogen heteroatoms in the carbon framework based on their location (Table S2†), pyridinic and pyrrolic N usually occurs at the domain edge. In another words, too much pyridinic and pyrrolic N introduced into the carbon framework will inevitably lead to the formation of vacancy defects and thus generate an additional hurdle for electron transport, which is unwanted for their application in air electrodes.26–29 Graphitic nitrogen dopants could simultaneously facilitate the electron transfer and “activate” adjacent C atoms and are thus preferred for electrochemical applications.1,6,30 However, there is still a lack of efficient methods to precisely insert nitrogen atoms into a carbon lattice merely at graphitic sites.
Besides the chemical structure, specific surface area and porous structure of nanocarbons determine the accessible parts of active sites and the mass transfer to absorb or release oxygen gas bubbles for the ORR and OER respectively. Template-based methods can be especially feasible and advantageous for preparing mesoporous N-doped catalysts with high specific surface areas by rationally selecting either soft or hard nanotemplates,31–33 whereas effective approaches for generating micropores in N-doped nanocarbons are rather limited.7,32 The carbon-based metal-free electrocatalysts reported so far cannot meet all of the requirements of efficient, rechargeable and durable air electrodes for constructing two-electrode metal–air batteries.
Herein, we report a facile method to control both the pore dimensions and the graphitic nitrogen doping of a net-like carbon nanomaterial via self-assembly and pyrolysis of nanoparticles and the commercial block copolymer poly(ethylene oxide)-block-poly(propylene oxide)-block-poly(ethylene oxide) (PEO20–PPO70–PEO20, P123). The graphitic nitrogen-doped carbon nano-nets with both micropores and mesopores, denoted g-N-MM-Cnet, performed as an excellent metal-free electrocatalyst for the reversible reduction and evolution of oxygen. Such a metal-free electrocatalyst could fulfill the requirements of reversible and durable air electrodes for two-electrode metal–air secondary batteries (exemplified with an alkaline zinc–air battery here).
2 Experimental section
2.1 Electrocatalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.0135:0.52:16:40. Finally, 2.8 g of dicyandiamide (DCDA) was added to the as-formed dispersion and dried at 40 °C overnight. The dried white powders were heated at 5 °C min−1 to 1000 °C under the protection of an N2 flow and kept at that temperature for 1 h. After cooling to room temperature, the as-formed black powder was etched with an excess amount of HF solution (20%) to remove the TiN and TiO2 components. After the etching process, the sample was further washed with water and ethanol and dried at 60 °C overnight. The g-N-MM-Cnet powder was used for further characterization and electrochemical tests. For more experimental details, please see the ESI (Table S1†).:0.52:16:40) and dried at 40 °C for 12 h. The as-obtained white powder was heated at 5 °C min−1 to 1000 °C under the protection of an N2 flow and kept at that temperature for 1 h. After naturally cooling to room temperature, the as-formed carbon sample was directly used for further characterization and electrochemical tests.
:0.0135:0.52:16:40. Finally, 2.8 g of dicyandiamide (DCDA) was added to the as-formed dispersion and dried at 40 °C overnight. The dried white powders were heated at 5 °C min−1 to 1000 °C under the protection of an N2 flow and kept at that temperature for 1 h. After cooling to room temperature, the as-formed black powder was etched with an excess amount of HF solution (20%) to remove the TiN and TiO2 components. After the etching process, the sample was further washed with water and ethanol and dried at 60 °C overnight. The g-N-MM-Cnet powder was used for further characterization and electrochemical tests. For more experimental details, please see the ESI (Table S1†).:0.52:16:40) and dried at 40 °C for 12 h. The as-obtained white powder was heated at 5 °C min−1 to 1000 °C under the protection of an N2 flow and kept at that temperature for 1 h. After naturally cooling to room temperature, the as-formed carbon sample was directly used for further characterization and electrochemical tests.For comparison, commercial Pt/C (20 wt% Pt) was used as received. Ir/C (20 wt% Ir) was prepared by an incipient wetness method according to the previous report.34 Vulcan XC 72R (100 mg) was impregnated with the appropriate amount of iridium which had previously been dissolved in deionized water. After stirring for 12 h, the impregnated powder was frozen in liquid nitrogen and freeze-dried under vacuum. The dried powder was heated to 465 °C at 10 °C min−1 in a tube furnace under a reductive atmosphere (5 vol% H2, 95 vol% Ar) and then kept at that temperature for 2 h.
2.2 Electrochemical measurements
The electrochemical activity was tested in a conventional three-electrode cell reactor by using the rotating disc electrode (RDE) technique at room temperature. A saturated calomel electrode and Ag/AgCl were used as reference electrodes in 0.1 M KOH and 0.5 M H2SO4 electrolyte solutions, respectively. A platinum net was used as the counter electrode in all electrolytes. A RDE with a glassy carbon disk (diameter: 5.0 mm, from PINE Instruments, USA) served as the substrate for the working electrode. The working electrode was prepared as follows: 5 mg of catalyst was mixed with 50 μl Nafion solution (5 wt% Nafion solution in alcohol) and 0.8 ml H2O in an ultrasonic bath to obtain a well dispersed ink. Then, a 10 μl aliquot of the catalyst ink was pasted on the glassy carbon surface. The ORR and OER activities of the catalysts were measured in O2-saturated 0.1 M KOH and 0.5 M H2SO4 with a sweep rate of 10 mV s−1 at 1600 rpm and 0 rpm respectively. The electrochemical impedance spectroscopy (EIS) measurements for the g-N-MM-Cnet and layered carbon were performed in an O2-saturated electrolyte with the frequency range of 100 kHz to 0.01 Hz. All measurements were conducted at room temperature. For comparison, 20 wt% Pt/C and Ir/C (20 wt% Ir) were measured using the same methods.The overall electron transfer number per oxygen molecule involved in a typical ORR process can be calculated from the slope of the Koutecky–Levich plots according to our previous work.35
The impedance properties of Cnet-800 °C, g-N-MM-Cnet and layered carbon were also evaluated using a three-electrode cell configuration. The working electrode was fabricated by coating an aliquot of the catalyst ink on the glassy carbon surface and testing at 0.85 V vs. RHE with a 5 mV AC potential from 30 kHz to 0.1 Hz. A saturated calomel electrode (SCE) was used as the reference electrode and a platinum net as the counter electrode. The electrolyte was O2-saturated 0.1 M KOH aqueous solution.
The conductivity of Cnet-800 °C, g-N-MM-Cnet and layered carbon was evaluated using a two-electrode cell configuration. The working electrode was fabricated using the following steps: 5 mg sample (90 wt%) and polytetrafluoroethylene (PTFE, 10 wt%) were well mixed and ground in an agate mortar and then pressed onto foam Ni. A platinum net was used as the counter electrode and reference electrode. The electrochemical impedance spectroscopy (EIS) measurements for g-N-MM-Cnet and layered carbon were performed in 0.1 M KOH solution at −1.5 V with a 5 mV AC potential from 100 kHz to 0.01 Hz.
For the Zn–air battery test, the air electrode was prepared by coating the as-prepared catalyst ink onto carbon paper (HCP 135) then drying at 45 °C for 3 h. The mass loading was 2 mg cm−2 unless otherwise noted. A Zn plate (0.5 mm) was used as the anode and a 6 M KOH solution containing 0.2 M zinc acetate was utilized as the electrolyte. The rechargeable Zn–air battery was assembled and tested in a home-made reactor.
2.3 Material characterizations
X-ray diffraction (XRD) patterns were obtained by X-ray diffraction (Rigaku D/Max-2550, Cu Kα λ = 1.54056 Å) measurement. Scanning electron microscopy (SEM) measurements were performed on a FEI Nova NanoSEM2300. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) measurements were taken with a JEM-2100F microscope operating at 200 kV accelerating voltage. X-ray photoemission spectroscopy (XPS) analysis was performed on a Kratos Axis Ultra DLD spectrometer with Al Kα radiation source. An inVia-reflex micro-Raman spectrometer (Renishaw, UK) was used to investigate the g-N-MM-Cnet and layered carbon samples with a 532 nm wavelength incident laser. A NOVA2200e analyzer (Quantachrome, USA) was used for porosity analysis. Samples were degassed at 250 °C for 12 h before measurements.3 Results and discussion
3.1 Synthesis of g-N-MM-Cnet
The self-assembly of Pluronic P123 could offer a powerful tool for constructing micelles, lamellar sheets and even lyotropic phases. As a result, such an amphiphilic block copolymer is usually applied as a typical soft template or structure directing reagent for casting inorganic replicates, whose morphology could be controlled at the nanoscale by solvents, chemical additives and temperature. In principle, the assembly of block copolymers could also be affected by inorganic species via strong interactions at the surface or interface. As depicted in Fig. 1, we developed a synthetic approach for fabricating g-N-MM-Cnet by carbonization of supramolecular entities of P123 with the assistance of TiO2 nanoparticles as multifunctional structure directing templates. | ||
| Fig. 1 Proposed process for the formation of the net-like structure in g-N-MM-Cnet: (a) assembly of P123 directed by in situ formed TiO2 nanoparticles from the hydrolysis of titanium tetraisopropoxide in an HCl solution of water and ethanol; (b) addition of dicyandiamide (DCDA) and removal of solvents; (c) carbonization of the as-obtained supramolecular entity at high temperature (optimized at 1000 °C) under N2 protection; (d) removal of Ti-containing species via chemical etching to release hierarchical pores. | ||
Large-area SEM images (Fig. 2a and S1†) could offer an overview of the hierarchical structure of g-N-MM-Cnet with a flower-like shape and thus, macroscale voids. Each “petal” in g-N-MM-Cnet was found to be composed of three-dimensional interconnected carbon networks as clearly seen in the amplified SEM image (Fig. 2b) with nanoscale “net” holes. HRTEM images further indicated that the subunits of g-N-MM-Cnet are ultra-fine graphite nanowires with a mean diameter of less than 3 nm (Fig. 2c). The XRD spectra of g-N-MM-Cnet (Fig. S3†) with typical peaks at 25.71 and 43.43 degrees rather suggested well-developed graphitized carbon domains, which can be further confirmed by the sharp G-band at 1594 cm−1 in the Raman spectra of g-N-MM-Cnet (Fig. S4†).36
 | ||
| Fig. 2 SEM images (a and b) and HRTEM image (c) of g-N-MM-Cnet. (d) Elemental mapping results of a typical carbon nano-net sample. | ||
The multi-scale pore dimensions of g-N-MM-Cnet were further demonstrated by the N2 adsorption–desorption analysis results. The g-N-MM-Cnet has a specific surface area of up to 1947 m2 g−1 and a pore volume of 2.67 cm3 g−1 with micropores centered at 0.64 nm and average bimodal mesopores centered at 5.66 nm (Fig. 3 and Table S3†). The micropore volume of g-N-MM-Cnet is 0.58 cm3 g−1. The control sample obtained without adding TTIP was found to be carbon nanoflakes (Fig. S2†) with a rather small surface area (106.8 m2 g−1) and negligible micropore volume (0.03 cm3 g−1). Both the micropore volume and micropore surface area of g-N-MM-Cnet are among the highest values for carbon materials in the literature.
 | ||
| Fig. 3 N2 adsorption/desorption isotherms (a) and pore size distribution curves (b) of g-N-MM-Cnet and layered carbon. | ||
The homogeneous distribution of N heteroatoms in the carbon framework was directly demonstrated by elemental mapping images (Fig. 2d). Further XPS analysis results (Fig. S5 and Table S4†) showed merely C, N and O elements present in g-N-MM-Cnet with a N content of 2.08 at%. The high-resolution N 1s XPS spectrum (Fig. 4 and S5 and Table S5†) of g-N-MM-Cnet with an energy peak at 401.1 eV revealed that all of the nitrogen heteroatoms located at graphitic sites. There is a consensus that the graphitic nitrogen in nanocarbons would benefit their potential applications as metal-free electrocatalysts via enriching active centers and elevation electron mobility.29 It is generally accepted in the literature that the percentage of graphitic nitrogen content among all types of nitrogen dopants will increase with synthetic temperature and can reach as high as 70% in some special cases.7,37 But success in precisely doping nanocarbons with graphitic nitrogen atoms has never been touched before this work. Note that only 82.93% of nitrogen heteroatoms in layered carbon are located at graphitic sites.
 | ||
| Fig. 4 High-resolution N 1s XPS spectra of g-N-MM-Cnet (a) and layered carbon (b) with typical peaks of the graphitic N and/or pyridinic N sites. | ||
3.2 Formation mechanism of g-N-MM-Cnet
The formation of net-like nanocarbons was sensitive to the amounts of P123, DCDA and TTIP involved. The yield of g-N-MM-Cnet increased with more P123 added (Fig. S6 and S7†), rather suggesting the role of P123 as a carbon source. As an oxygen rich polymer, P123 is usually used as a removable ligand to get mesoporous mineral nanomaterials. In order to ensure the carbonization of P123 with high yields, DCDA was used as an efficient reductant to reduce the oxygen-containing species for maintaining the nanostructures of the P123 assemblies (Fig. S8 and S9†). Direct condensation of DCDA at around 550 °C could give graphitic carbon nitride, which decomposed into gaseous molecules completely at a temperature higher than 700 °C (Fig. S10†). As a result, the redox reaction between P123 and DCDA is of key importance for the formation of carbon nano-nets with acceptable yields (calculated on the basis of the amount of P123 involved). Moreover, as the only nitrogen-containing precursor, DCDA also functioned as the nitrogen source for the formation of TiN and nitrogen-doped carbon samples (Fig. S8 and S9†). The molecular ratio of TTIP:P123:DCDA was finally optimized to be 1:0.0135:2.5 for preparing g-N-MM-Cnet with relatively high yields.
The involvement of TTIP and thus TiO2 nanoparticles dominated the formation of a net-like structure (Fig. S1 and S2,† 2a and b) revealing their role as hard-templates for directing the assembly of P123. Only large-area layered carbon formed when TTIP and thus TiO2 nanoparticles were excluded from the synthetic system. XRD analysis results suggested a transformation process of TiO2 into TiN components from 600 to 800 °C (Fig. 5a). The nitrogen dopants (mainly graphitic and pyridinic nitrogen atoms) in the carbon frameworks should act as a possible nitrogen source for further growth of TiN crystals. TiN nanocrystals simultaneously grew up into larger ones, as reflected by their ever stronger and sharper XRD peaks (Fig. 5a) of TiN species at elevated temperatures from 800 to 1000 °C. Moreover, g-N-MM-Cnet had a much lower nitrogen content than the control sample of layered carbon (Fig. 4 and S6 and Table S4†). Only graphitic nitrogen dopants in g-N-MM-Cnet were obtained at 1000 °C (Fig. 5b, S13 and S14 and Table S5†). Such a chemical removal of pyridinic nitrogen-containing complexes could simultaneously lead to a slight shrinkage in the frameworks of the carbon net with gradually enlarged mesopores (Fig. 5c and S15†) and increased pore volumes of both mesopores and micropores (Table S3†) in g-N-MM-Cnet. All of these results rather suggested the important role of TiO2 species as selective “absorbers” of nitrogen atoms at the edge or defect sites to prepare graphitic nitrogen-doped and microporous nanocarbons (Fig. 5e).
 | ||
| Fig. 5 (a) XRD patterns of TiN/Cnet prepared at different temperatures. High-resolution N 1s spectrum (b), pore size distribution (c) and (d) of Cnet samples obtained at different temperatures after the removal of TiN templates. (e) Proposed reaction path between TiO2 species and nitrogen-containing complexes for the formation of microporous g-N-MM-Cnet: C black, N green, TiO2 blue and TiN yellow. Only a monolayer of graphite is presented in (d) for better clarification. | ||
3.3 ORR and OER activities of g-N-MM-Cnet
High surface area, hierarchical nanopores from micropores to mesopores and graphitic nitrogen dopants, all of these merits make g-N-MM-Cnet an excellent candidate for potential applications as ORR and/or OER electrocatalysts. Indeed, the g-N-MM-Cnet electrode affords an onset potential of 0.96 V versus the reversible hydrogen electrode (RHE), surpassing the Pt/C electrode (onset potential = 0.94 V). The half-wave potential (E1/2) of the g-N-MM-Cnet electrode (E1/2 = 0.88 V) was 40 mV higher than that of Pt/C (E1/2 = 0.84 V). Layered carbon and Cnet samples obtained at the same temperature with lower surface area and more or less pyridinic nitrogen dopants all performed in the ORR even worse than Pt/C (Fig. 6a and S18†). A similar trend of ORR activities was also observed in acid electrolytes for g-N-MM-Cnet and the control carbon samples (Fig. S20 and S21†). The much higher E1/2 and thus working potentials of g-N-MM-Cnet are preferred for their practical applications in fuel cells and air batteries to ensure high energy conversion efficiency. | ||
| Fig. 6 ORR and OER polarization curves (a), electron transfer number (b) and diffusion-current-corrected OER Tafel plots (c) of various catalysts on a RDE in 0.1 M KOH. The loading was 0.6 mg cm−2 for all materials. Inset: the diffusion-current-corrected Tafel plots of g-N-MM-Cnet and Pt/C for ORR. (d) ORR and OER polarization curves of g-N-MM-Cnet and Pt/C on Teflon-coated carbon fiber paper (CFP) electrodes in 6 M KOH. The catalyst loading on CFP electrodes was 2 mg cm−2 for all materials. | ||
The electron-transfer number values of the g-N-MM-Cnet electrocatalyst were estimated to be 3.91–3.9 from 0.7 to 0.1 V versus RHE (Fig. 6b), suggesting a favorable 4e pathway of the ORR with water as the main product (Fig. S19†). The Tafel slope of 70 mV per decade (in 0.1 M KOH) for g-N-MM-Cnet (Fig. 6b inset) was close to that of Pt/C (64 mV per decade). The g-N-MM-Cnet sample also shows much better electrochemical stability in both acidic and basic medium (Fig. S22†). For direct use in methanol fuel cells, ORR catalysts should also exhibit a satisfactory tolerance to methanol that may pass across the membrane from the anode. As shown in Fig. S22,† the original cathodic ORR current of g-N-MM-Cnet was not obviously depressed by the crossover effect, whilst Pt/C and layered carbon exhibited a distinct methanol oxidation reaction after the introduction of methanol (Fig. S22c†). Indeed, g-N-MM-Cnet is an ideal cathode catalyst for a direct methanol alkaline fuel cell.
The g-N-MM-Cnet sample also exhibited excellent OER activity (Fig. 6a and c, S18 and S21†) in both acidic and alkaline electrolytes. For example, the g-N-MM-Cnet offered overpotentials down to 0.37 V and 0.15 V vs. RHE for the OER in 0.1 M KOH and 0.5 M H2SO4 respectively at a current density (J) of 10 mA cm−2, which was more negative than Pt/C and other control samples. The involvement of pyridinic nitrogen dopants obviously increased the overpotentials for the OER (Fig. S18† and 6a). The onset potentials of these samples for the OER in the sequence g-N-MM-Cnet ∼ N-MM-Cnet-900 ∼ N-MM-Cnet-800 ≪ layered carbon also suggested the key role of microporous structure in significantly boosting the OER activity of N-MM-Cnet. A more favourable OER reaction over g-N-MM-Cnet was further reflected by comparing the Tafel slope to the state-of-the-art Ir/C catalyst (Fig. 6c). Nitrogen dopants can supply possible active sites for electrocatalysts. However, pyridinic N and pyrrolic N will inevitably lead to the formation of vacancy defects and thus hinder charge transfer, which is harmful towards improving the activity of carbon electrocatalysts. Only graphitic nitrogen exists in g-N-MM-Cnet which could “activate” adjacent C atoms and simultaneously facilitate the electron transfer (Fig. S23†), resulting in much better ORR and OER activity over g-N-MM-Cnet. The diffusion resistance (Fig. S23a†) of the g-N-MM-Cnet-based work electrode was much lower than those of the control samples due to the microporous structure and high conductivity of the carbon frameworks. Notably, both ORR and OER activity of g-N-MM-Cnet on CFP were further improved in 1 M (Fig. S25†) and 6 M (Fig. 6d) KOH electrolytes, promising the great potential of g-N-MM-Cnet-based air electrodes for constructing Zn–air secondary batteries.
3.4 Rechargeable Zn–air batteries
After discovering the excellent ORR and OER activities of g-N-MM-Cnet, we naturally extended the application of g-N-MM-Cnet as an air electrode in a Zn–air battery (Fig. 7). The g-N-MM-Cnet air electrode showed an open circuit voltage of around 1.28 V, a current density of 560 mA cm−2 and a peak power of 324 mW cm−2 at 0.624 V. Both the current density at 1 V and peak power density were significantly improved over previous reports on Zn–air batteries made of noble-metal-free electrodes (Table S6†),16,18,19 or Pt/C air cathodes with the same catalyst loading here (Fig. 7b). The g-N-MM-Cnet-based Zn–air battery delivered a specific capacity of 667.8 mA h kgZn−1 with an energy density of up to 866.4 W h kgZn−1 upon full discharging at 5 mA cm−2 (Fig. 7c), comparable to or slightly higher than the energy densities of Pt/C (Fig. S26†) and the best non-noble metal-based zinc–air batteries reported in the literature (Table S6†). The energy density could be maintained as high as 815.3 and 774.1 W h kgZn−1 upon full discharging at 20 and 40 mA cm−2, respectively, indicating very low loss in the energy intensity and specific capacity (Fig. 7c) of the g-N-MM-Cnet-based Zn–air battery at higher discharge rates.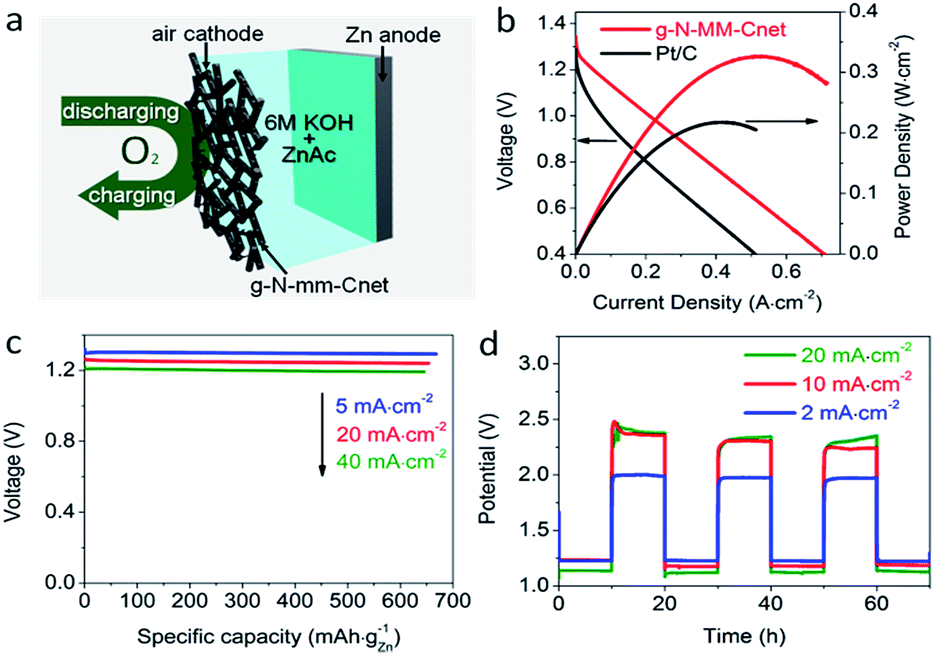 | ||
| Fig. 7 (a) A schematic of the g-N-MM-Cnet-based two-electrode Zn–air battery. (b) Polarization curves (V–i) and corresponding power plots of two-electrode Zn–air batteries made of g-N-MM-Cnet and Pt/C air cathodes. (c) Long-time discharge curves of a g-N-MM-Cnet-based two-electrode Zn–air battery tested at 5, 20 and 40 mA cm−2, giving specific capacities of 667.8, 653.7 and 645.2 mA h kgZn−1, respectively. (d) Discharge/charge cycling curves of the two-electrode rechargeable Zn–air battery at 20, 10 and 2 mA cm−2 respectively in a 20 h cycle period. | ||
The g-N-MM-Cnet-based Zn–air battery also showed excellent rechargeability (Fig. 7d). The charge–discharge voltage gap of the g-N-MM-Cnet electrode was smaller than that of Pt/C and comparable to that of the Pt/C + Ir/C electrodes at lower current density and outperformed all these control electrodes at 50 mA cm−2 or higher (Fig. S27†). Note that the voltage polarization was mainly contributed by the sum of the OER and ORR overpotentials at the air cathode (Fig. S28†), whilst contribution of the Zn anode was negligible in our devices. When the g-N-MM-Cnet-based Zn–air battery was repeatedly charged or discharged at 20 mA cm−2, 10 mA cm−2 and 2 mA cm−2 over a 20 h per cycle period, the battery showed high cycling stability (Fig. 7d).
3.5 Durability of two-electrode Zn–air batteries
The durable performance of the g-N-MM-Cnet-based two-electrode Zn–air battery was also well reflected by 1475 discharge/charge cycles at 2 mA cm−2 in a 1200 s cycle period with an even smaller charge–discharge voltage gap over a period of 491 h (Fig. 8), outperforming all state-of-the-art rechargeable Zn–air batteries reported in the literature (Table S7†),6,14 including both two-electrode and three-electrode batteries. Although the charge–discharge voltage gap of Pt/C + Ir/C electrode was only 0.56 V at 2 mA cm−2, slightly smaller than the voltage gap of the g-N-MM-Cnet electrode (0.8 V at the first cycle), the charge–discharge voltage gap became broader after multiple charging–discharging cycles reaching 0.90 V after 80 cycles (Fig. S29†). | ||
| Fig. 8 Discharge/charge cycling curve of the g-N-MM-Cnet-based two-electrode Zn–air battery at 2 mA cm−2 over a 1200 s cycle period. The state-of-the-art lifetimes of two-electrode and three-electrode Zn–air batteries in previous work were also presented in purple and light purple, respectively. | ||
High durability of the g-N-MM-Cnet-based two-electrode Zn–air battery mainly originated from the high stability of the active centers for both the ORR and OER, that is, the graphitic nitrogen–carbon complexes. Pyridinic nitrogen–carbon complexes are also possible active centers for oxygen adsorption/desorption and, as a result, activation for the ORR.6–10 However, the stability of pyridinic nitrogen–carbon complexes for reversible ORR/OER processes is rather low presumably due to the oxidation of pyridinic nitrogens2 resulting in irreversible loss in the ORR active sites. Those well explain the excellent ORR activity of pyridinic nitrogen-rich carbon nanomaterials with low durability when merged in rechargeable two-electrode metal–air batteries. The excellent thermal stability of graphitic nitrogen atoms has already been demonstrated in the former section. It is thus reasonable that graphitic nitrogen–carbon complexes, as exemplified with g-N-MM-Cnet here, with an excellent balance between electrochemical activity (for both OER and ORR) and stability, exhibited an ultra-long lifetime as an air electrode for two-electrode zinc–air batteries.
4 Conclusions
In summary, three-dimensional g-N-MM-Cnet with multi-level porous morphology, obtained by self-assembly from Pluronic P123, and TiO2, was used as a hard-template and nitrogen atom sacrificial agent. The catalyst exhibits high ORR and OER activity and better stability in the alkaline electrolyte compared to that of Pt/C catalysts. Its activity also exceeds that of other non-precious metal catalysts, the g-N-MM-Cnet catalysts promised to replace Pt-based catalysts in alkaline media. Besides that, g-N-MM-Cnet shows efficient catalytic activity for both the OER and ORR. As a bifunctional air electrode, the g-N-MM-Cnet-based two-electrode Zn–air secondary battery exhibited high energy density and an ultra-long lifetime. Further optimization of the pore dimension and the heteroatom dopant concentration is also in progress for extending the wide applications of the unique g-N-MM-Cnet sample in catalysis, sensors and other energy storage devices in our lab.Acknowledgements
This work was supported by the National Basic Research Program of China (2013CB934102), the National Natural Science Foundation of China (21331004, 21301116), the SJTU-UM joint grant and the Shanghai Eastern Scholar Program.References
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- Y. G. Li, W. Zhou, H. L. Wang, L. M. Xie, Y. Y. Liang, F. Wei, J. C. Idrobo, S. J. Pennycook and H. J. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281–7285 CrossRef CAS PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390–2396 Search PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. S. Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
- J. T. Zhang, Z. H. Zhao, Z. H. Xia and L. M. Dai, Nat. Nanotechnol., 2015, 10, 444–453 CrossRef CAS PubMed.
- H. W. Liang, X. D. Zhuang, S. Brüller, X. L. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- J. Moorhouse, Modern Chlor-Alkali Technology, Wiley-Blackwell, London, 2001, vol. 8 Search PubMed.
- T. N. Ye, L. B. Lv, X. H. Li, M. Xu and J. S. Chen, Angew. Chem., Int. Ed., 2014, 53, 6905–6909 CrossRef CAS PubMed.
- L. Wang, P. Yu, L. Zhao, C. G. Tian, D. D. Zhao, W. Zhou, J. Yin, R. H. Wang and H. G. Fu, Sci. Rep., 2014, 4, 5184–5192 CAS.
- X. R. Wang, X. L. Li, L. Zhang, Y. K. Yoon, P. K. Weber, H. L. Wang, J. Guo and H. J. Dai, Science, 2009, 324, 768–771 CrossRef CAS PubMed.
- Z. L. Wang, D. Xu, J. J. Xu and X. B. Zhang, Chem. Soc. Rev., 2014, 43, 7746–7786 RSC.
- S. J. Yang, M. Antonietti and N. Fechler, J. Am. Chem. Soc., 2015, 137, 8269–8273 CrossRef CAS PubMed.
- Y. G. Li, M. Gong, Y. Y. Liang, J. Feng, J. E. Kim, H. L. Wang, G. S. Hong, B. Zhang and H. J. Dai, Nat. Commun., 2013, 4, 1805–1812 CrossRef PubMed.
- T. Y. Ma, J. R. Ran, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 4646–4650 CrossRef CAS PubMed.
- V. M. Dhavale and S. Kurungot, ACS Catal., 2015, 5, 1445–1452 CrossRef CAS.
- K. N. Jung, S. M. Hwang, M. S. Park, K. J. Kim, J. G. Kim, S. X. Dou, J. H. Kim and J. W. Lee, Sci. Rep., 2014, 5, 7665–7675 CrossRef PubMed.
- L. Xu, Z. M. Luo, Z. X. Fan, X. Zhang, C. L. Tan, H. Li, H. Zhang and C. Xue, Nanoscale, 2014, 6, 11738–11743 RSC.
- W. H. He, C. H. Jiang, J. B. Wang and L. H. Lu, Angew. Chem., Int. Ed., 2014, 53, 9503–9507 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 22, 443–447 CrossRef PubMed.
- P. Zhang, Z. Qiao, Z. Zhang, S. Wan and S. Dai, J. Mater. Chem. A, 2014, 2, 12262–12269 CAS.
- F. Gao, G. L. Zhao, S. Z. Yang and J. J. Spivey, J. Am. Chem. Soc., 2013, 135, 3315–3318 CrossRef CAS PubMed.
- X. H. Li and M. Antonietti, Angew. Chem., Int. Ed., 2013, 52, 4572–4576 CrossRef CAS PubMed.
- Z. L. Li, J. H. Liu, Z. W. Huang, Y. Yang, C. G. Xia and F. W. Li, ACS Catal., 2013, 3, 839–845 CrossRef CAS.
- Y. H. Xue, J. Liu, H. Chen, R. G. Wang, D. Q. Li, J. Qu and L. M. Dai, Angew. Chem., Int. Ed., 2012, 51, 12124–12127 CrossRef CAS PubMed.
- B. Kumar, M. Asadi, D. Pisasale, S. Sinha-Ray, B. Rosen, R. Haasch, J. Abiade, A. L. Yarin and A. Salehi-Khojin, Nat. Commun., 2013, 4, 2819–2827 Search PubMed.
- H. B. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- Y. Zhao, C. G. Hu, Y. Hu, H. H. Cheng, G. Q. Shi and L. T. Qu, Angew. Chem., Int. Ed., 2012, 51, 11371–11375 CrossRef CAS PubMed.
- T. Schiros, D. Nordlund, L. Pálová, D. Prezzi, L. Y. Zhao, K. S. Kim, U. Wurstbauer, C. Gutiérrez, D. Delongchamp, C. Jaye, D. Fischer, H. Ogasawara, L. G. M. Pettersson, D. R. Reichman, P. Kim, M. S. Hybertsen and A. N. Pasupathy, Nano Lett., 2012, 12, 4025–4031 CrossRef CAS PubMed.
- Y. J. Gao, G. Hu, J. Zhong, Z. J. Shi, Y. S. Zhu, D. S. Su, J. G. Wang, X. H. Bao and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 2109–2113 CrossRef CAS PubMed.
- X. H. Li, S. Kurash, U. Kaiser and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 9689–9692 CrossRef CAS PubMed.
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed.
- J. Kim, C. Anand, S. N. Talapaneni, J. You, S. S. Aldeyab, E. Kim and A. Vinu, Angew. Chem., Int. Ed., 2012, 51, 2859–2863 CrossRef CAS PubMed.
- T. Reier and M. Oezaslan, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- L. B. Lv, T. N. Ye, L. H. Gong, K. X. Wang, J. Su, X. H. Li and J. S. Chen, Chem. Mater., 2015, 27, 544–549 CrossRef CAS.
- Q. Q. Zhuo, Q. Yang, Y. P. Zhang, D. Q. Zhang, L. Li, C. H. Gao, Y. Q. Sun, L. Ding, Q. J. Sun, S. D. Wang, J. Zhong, X. H. Sun and S. T. Lee, ACS Nano, 2015, 9, 594–601 CrossRef CAS PubMed.
- W. W. Lei, D. Portehault, R. Dimova and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 7121–7127 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra08815e |
| This journal is © The Royal Society of Chemistry 2016 |