
Polycyclic polyprenylated acylphloroglucinols: natural phosphodiesterase-4 inhibitors from Hypericum sampsonii†
Jun-Sheng Zhang,
Yi-Hong Zou,
Yan-Qiong Guo,
Zhen-Zhen Li,
Gui-Hua Tang and
Sheng Yin*
School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou, Guangdong 510006, P. R. China. E-mail: yinsh2@mail.sysu.edu.cn; Fax: +86-20-39943090; Tel: +86-20-39943090
First published on 26th May 2016
Abstract
Chemical investigation of the aerial parts of Hypericum sampsonii led to the isolation of seven new polycyclic polyprenylated acylphloroglucinols, hypersampsonones A–G (1–7), together with 23 known analogs (8–30). Their structures including the absolute configurations were elucidated by combined spectroscopic analysis, quantum chemical ECD calculations, and chemical methods. Compound 1 represents an unprecedented cyclocitral monoterpene-coupled bicyclo[3.3.1]nonane skeleton, while 2 features an unusual hexahydrofuro[2,3-b]furan-diepoxy ring system fused in a tricyclo[4.3.1.15,7]undecane skeleton. All the compounds were screened by using tritium-labeled adenosine 3′,5′-cyclic monophosphate ([3H]-cAMP) as substrate for their inhibitory activity against phosphodiesterase-4 (PDE4), which is a drug target for the treatment of asthma and chronic obstructive pulmonary disease. Compounds 1, 18–19, 21, and 25–30 exhibited inhibition with IC50 values less than 10 μM, in which compound 19 represented the most active compound (IC50 = 0.64 μM), being comparable to the positive control, rolipram (IC50 = 0.62 μM).
Introduction
Polycyclic polyprenylated acylphloroglucinols (PPAPs) are a class of hybrid natural products characterized by a highly oxygenated and densely substituted bicyclo[3.3.1]nonane-2,4,9-trione or other related core structures. PPAPs are biosynthetically generated through a “mixed” mevalonate/methylerythritol phosphate and polyketide pathway and are usually decorated with prenyl or geranyl side chains.1,2 Over the past decades, more than 300 structurally diverse PPAPs have been reported,1,2 and their fascinating highly rigid “diamond-like” architectures as well as important biological activities have attracted broad interest from both natural products and synthetic chemists.3–6 Hyperforin, for instance, has been reported to possess a wide range of biological effects including antibiotic, anticancer, antidepressant, and procognitive activities, and has been totally synthesized by five research groups within the last five years.7,8 So far, natural PPAPs have been reported exclusively from plants of the Guttiferae family, and nearly half of these PPAPs were isolated from the genus Hypericum.1,2 Particularly, Hypericum sampsonii, an evergreen shrub used in traditional Chinese medicine for removing blood stasis and relieving swelling,9 has been proved to be a rich source of complex caged PPAPs with a wide variety of biological properties, such as antimicrobial,10 cytotoxic,5 and anti-HIV11 activities.Phosphodiesterase-4 (PDE4), which specifically catalyzes the hydrolysis of adenosine 3′,5′-cyclic monophosphate (cAMP), is a promising drug target of high interest for central nervous system (CNS), inflammatory, and respiratory diseases.12 In our continuing search for PDE4 inhibitors from medicinal plants,13,14 seven new PPAPs, hypersampsonones A–H (1–7), and 23 analogues were isolated from the aerial parts of H. sampsonii. Compound 1 represents an unprecedented carbon skeleton formed from coupling of a cyclocitral monoterpene and a bicyclo[3.3.1]nonane, while 2 features an unusual hexahydrofuro[2,3-b]furan-diepoxy ring system fused in a tricyclo[4.3.1.15,7]undecane skeleton. All of the isolates were screened for their inhibitory activity against PDE4, and ten compounds were identified as PDE4 inhibitors with IC50 values less than 10 μM. Herein, the details of the isolation, structure elucidation, and inhibitory activity of these compounds are described.
Results and discussion
The air-dried powder of aerial parts of H. sampsonii was extracted with 95% EtOH at room temperature (rt) to give a crude extract, which was suspended in H2O and successively partitioned with petroleum ether (PE) and EtOAc. Various column chromatographic separations of the PE extract afforded compounds 1–30 (Fig. 1).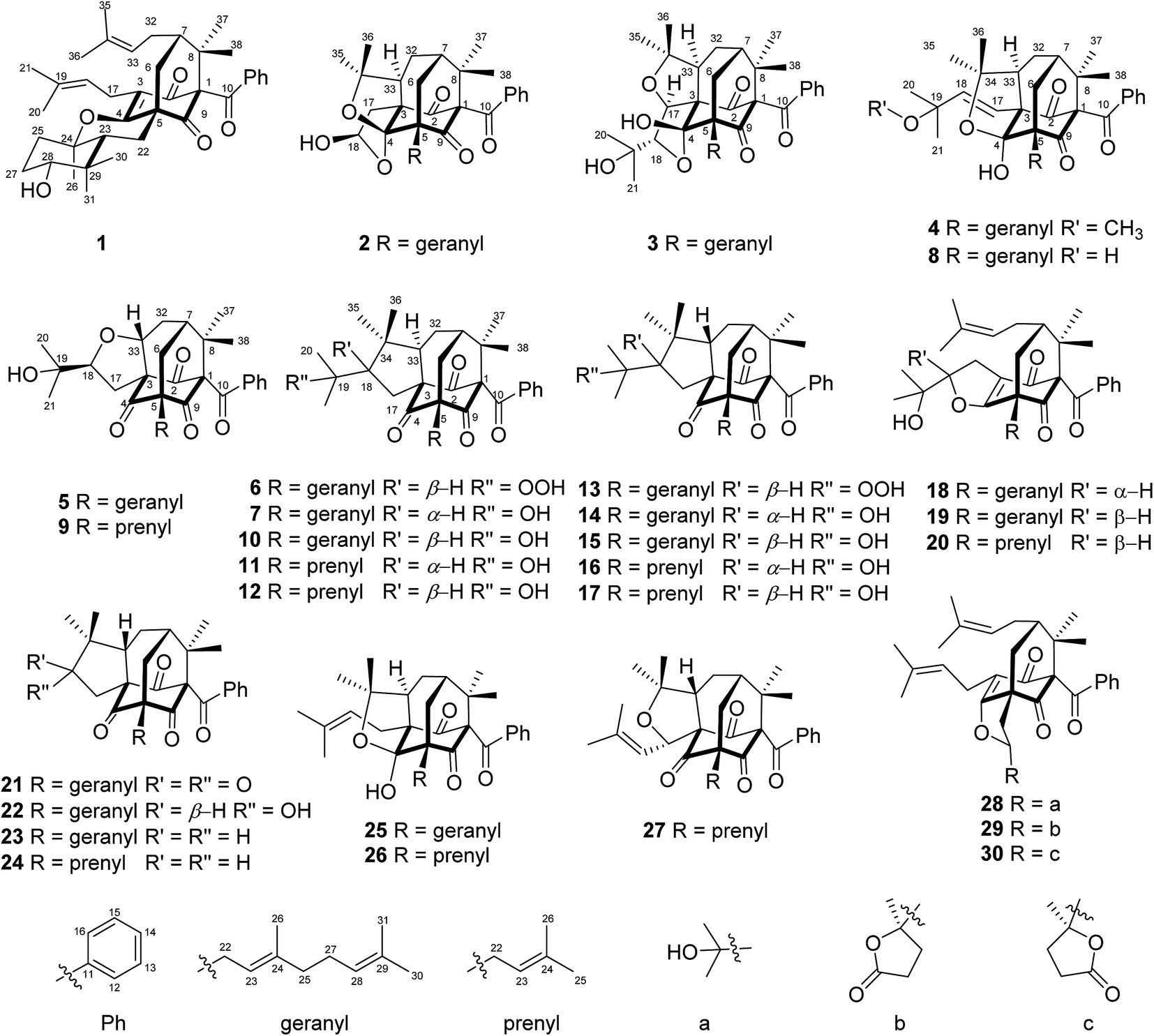 | ||
| Fig. 1 Structures of compounds 1–30. | ||
Compound 1, a colorless oil, had the molecular formula C38H50O5 as determined by the HRESIMS ion at m/z 585.3561 (calcd for [M − H]−, 585.3585), indicating 14 degrees of unsaturation (DOUs). The IR spectrum exhibited absorption bands for hydroxyl (3505 cm−1) and carbonyl (1721 and 1694 cm−1) functionalities. The 1H and 13C NMR data (Tables 1 and 2) and HSQC spectrum showed signals for a benzoyl [δH 7.22 (2H, brt, J = 7.5 Hz), 7.37 (1H, tt, J = 7.5, 1.2 Hz), and 7.48 (2H, brd, J = 7.5 Hz); δC 127.8 × 2, 128.2 × 2, 131.7, 137.0, and 193.8], an α,β-unsaturated ketone (δC 123.3, 167.6, and 192.6), a ketone (δC 206.2), and two prenyl groups [δH 1.54 (3H, s), 1.59 (3H, s), 1.61 (3H, s), 1.68 (3H, s), 2.13 (2H, m), 2.96 (1H, dd, J = 13.5, 7.1 Hz), 3.08 (1H, dd, J = 13.5, 7.7 Hz), 4.87 (1H, dd, J = 7.2, 6.0 Hz), 4.99 (1H, dd, J = 7.7, 7.1 Hz); δC 17.8, 18.0, 22.4, 25.8, 25.9, 29.0, 120.4, 124.8, 132.1, 132.4], suggesting that 1 was a PPAP derivative.10 The characteristic carbon signals for three sp3 quaternary carbons at δC 76.7 (C-1), δC 52.4 (C-5), and δC 48.2 (C-8), together with a sp3 methine at δC 47.7 (C-7) indicated that 1 possessed a bicyclo[3.3.1]nonane core, similar to that of propolone D.15 Comparison of the NMR data of 1 with those of propolone D indicated that their major differences arose from the C10 unit linked at C-5 of the bicyclo[3.3.1] core. As 12 of the 14 DOUs were accounted for by a benzoyl group, two keto groups, three double bonds, and a bicycle core, the remaining DOUs required two additional rings in the C10 unit.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| 6 | a 2.09, m; b 2.41, d (14.4) | a 2.23, m; b 2.35, m | a 2.27, m; b 2.45, m | 2.26, m | 1.99, m | a 1.89, m; b 2.51, m | a 1.84, m; b 2.51, m |
| 7 | 1.46, m | 1.78, m | 2.03, m | 1.73, m | 2.00, m | 2.11, m | 2.14, m |
| 12 | 7.48, brd (7.5) | 7.57, a (7.5) | 7.58, brt (7.4) | 7.44, brd (7.5) | 7.11, brd (7.1) | 7.10, brd (7.4) | 7.11, brd (7.4) |
| 13 | 7.22, brt (7.5) | 7.36, brt (7.5) | 7.26, brt (7.4) | 7.27, brt (7.5) | 7.31, brt (7.1) | 7.28, brt (7.4) | 7.26, brt (7.4) |
| 14 | 7.37, tt (7.5, 1.2) | 7.39, brt (7.5) | 7.38, brt (7.4) | 7.35, brt (7.5) | 7.42, brt (7.1) | 7.39, brt (7.4) | 7.39, brt (7.4) |
| 15 | 7.22, brt (7.5) | 7.36, brt (7.5) | 7.26, brt (7.4) | 7.27, brt (7.5) | 7.31, brt (7.1) | 7.28, brt (7.4) | 7.26, brt (7.4) |
| 16 | 7.48, brd (7.5) | 7.57, brd (7.5) | 7.58, brt (7.4) | 7.44, brd (7.5) | 7.11, brd (7.1) | 7.10, brd (7.4) | 7.11, brd (7.4) |
| 17 | a 2.96, dd (13.5, 7.1); b 3.08, dd (13.5, 7.7) | a 2.45, m; b 2.96, m | 5.08, brs | 6.44, d (16.5) | a 2.01, m; b 3.04, m | a 2.17, m; b 2.49, m | a 2.43, dd (9.5, 13.5); b 2.91, dd (11.4, 13.5) |
| 18 | 4.99, dd (7.7, 7.1) | 5.86, m | 4.00, brs | 5.90, d (16.5) | 4.30, dd (7.2, 7.2) | 2.84, dd (7.9, 12.3) | 1.95, m |
| 20 | 1.61, s | 1.28, s | 1.17, s | 1.24, s | 1.35, s | 1.30, s | |
| 21 | 1.59, s | 1.33, s | 1.21, s | 1.16, s | 1.39, s | 1.38, s | |
| 22 | α 1.19, m; β 3.15, d (11.3) | a 2.45, m; b 2.76, dd (14.4, 7.2) | a 2.53, m; b 2.85, m | a 2.45, m; b 2.70, dd (6.9, 6.9) | 2.59, d (7.2) | 2.56, d (6.8) | 2.54, m |
| 23 | 1.14, m | 5.39, dd (7.2, 7.2) | 5.48, m | 5.52, dd (6.9, 6.9) | 5.23, dd (7.2, 7.2) | 5.13, dd (6.8, 6.8) | 5.01, m |
| 25 | α 2.12, m; β 1.67, m | 2.04, m | 2.10, m | 2.04, m | 2.03, m | 1.98, m | 1.95, m |
| 26 | 1.50, s | 1.65, s | 1.73, s | 1.68, s | 1.65, s | 1.65, s | 1.65, s |
| 27 | α 1.89, m; β 1.58, m | 2.03, m | 2.11, m | 2.05, m | 1.61, m | 1.99, m; 2.05, m | 1.96, m; 2.04, m |
| 28 | 3.37, dd (11.2, 4.3) | 5.07, dd (6.8, 6.8) | 5.06, m | 5.07, brs | 5.04, dd (7.2, 7.2) | 5.05, m | 5.04, m |
| 30 | 1.08, s | 1.58, s | 1.61, s | 1.59, s | 1.57, s | 1.58, s | 1.58, s |
| 31 | 0.82, s | 1.63, s | 1.69, s | 1.68, s | 1.65, s | 1.65, s | 1.66, s |
| 32 | 2.13, m | a 1.26, m; b 1.95, m | 2.07, m | 2.05, m | 2.10, m | 1.62, m | 1.49, m |
| 33 | 4.87, dd (7.2, 6.0) | 2.46, m | 2.50, m | 2.62, m | 4.21, dd (8.3, 8.3) | 2.05, m | 2.04, m |
| 35 | 1.68, s | 1.58, s | 1.35, s | 1.51, s | 0.92, s | 1.08, s | |
| 36 | 1.54, s | 1.47, s | 1.35, s | 1.44, s | 1.17, s | 0.98, s | |
| 37 | 1.35, s | 1.22, s | 1.48, s | 1.22, s | 1.40, s | 1.39, s | 1.44, s |
| 38 | 1.44, s | 1.42, s; 18-OH: 3.41, d (3.9) | 1.39, s; 4-OH: 4.35, s | 1.41, s; 19-OMe: 2.79, s | 1.42, s | 1.46, s | 1.50, s |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|
| 1 | 76.7 | 81.4 | 82.3 | 81.0 | 81.1 | 80.8 | 81.3 |
| 2 | 192.6 | 207.2 | 203.4 | 202.4 | 201.3 | 203.6 | 205.7 |
| 3 | 123.3 | 67.5 | 77.2 | 67.6 | 76.5 | 73.4 | 73.1 |
| 4 | 167.6 | 115.2 | 106.6 | 108.8 | 200.7 | 206.4 | 205.6 |
| 5 | 52.4 | 56.4 | 57.0 | 59.2 | 67.7 | 69.0 | 68.4 |
| 6 | 39.8 | 31.2 | 41.2 | 33.6 | 35.5 | 42.0 | 44.9 |
| 7 | 47.7 | 43.7 | 44.7 | 43.5 | 41.5 | 43.8 | 43.4 |
| 8 | 48.2 | 47.9 | 52.1 | 48.1 | 47.8 | 50.8 | 51.6 |
| 9 | 206.2 | 208.1 | 207.5 | 209.0 | 204.1 | 204.7 | 205.0 |
| 10 | 193.8 | 194.4 | 194.4 | 194.3 | 192.4 | 192.5 | 192.8 |
| 11 | 137.0 | 135.8 | 136.7 | 135.9 | 134.8 | 134.9 | 135.1 |
| 12 | 128.2 | 129.3 | 129.6 | 129.1 | 128.6 | 128.9 | 129.1 |
| 13 | 127.8 | 128.3 | 127.7 | 127.9 | 128.3 | 128.1 | 127.9 |
| 14 | 131.7 | 132.1 | 131.8 | 131.8 | 132.4 | 132.1 | 132.0 |
| 15 | 127.8 | 128.3 | 127.7 | 127.9 | 128.3 | 128.1 | 127.9 |
| 16 | 128.2 | 129.3 | 129.6 | 129.1 | 128.6 | 128.9 | 129.1 |
| 17 | 22.4 | 45.8 | 83.1 | 127.2 | 32.7 | 33.1 | 31.0 |
| 18 | 120.4 | 98.6 | 89.4 | 141.3 | 87.6 | 54.4 | 59.5 |
| 19 | 132.1 | 69.8 | 75.2 | 72.5 | 85.6 | 73.0 | |
| 20 | 25.8 | 27.3 | 24.4 | 26.7 | 24.7 | 31.0 | |
| 21 | 18.0 | 26.4 | 26.5 | 24.7 | 23.0 | 30.2 | |
| 22 | 22.9 | 27.3 | 28.5 | 28.8 | 28.9 | 29.3 | 29.9 |
| 23 | 48.7 | 119.8 | 120.0 | 120.2 | 118.4 | 119.0 | 119.0 |
| 24 | 82.7 | 137.9 | 140.4 | 139.3 | 139.3 | 138.4 | 138.4 |
| 25 | 38.9 | 40.0 | 40.0 | 40.0 | 40.0 | 40.0 | 39.9 |
| 26 | 21.8 | 16.2 | 16.3 | 16.2 | 16.3 | 16.4 | 16.4 |
| 27 | 28.5 | 26.7 | 26.3 | 25.8 | 26.5 | 26.6 | 26.6 |
| 28 | 77.4 | 124.3 | 123.8 | 124.0 | 124.0 | 124.1 | 124.1 |
| 29 | 38.8 | 131.4 | 131.9 | 131.7 | 131.5 | 131.4 | 131.3 |
| 30 | 27.3 | 17.7 | 17.7 | 17.7 | 17.7 | 17.7 | 17.7 |
| 31 | 14.3 | 25.7 | 25.8 | 25.7 | 25.7 | 25.7 | 25.7 |
| 32 | 29.0 | 25.3 | 28.9 | 26.5 | 31.3 | 28.6 | 28.3 |
| 33 | 124.8 | 49.4 | 50.1 | 52.7 | 81.6 | 57.8 | 58.4 |
| 34 | 132.4 | 84.2 | 86.8 | 84.7 | 44.5 | 47.2 | |
| 35 | 25.9 | 30.4 | 32.0 | 33.2 | 28.0 | 30.3 | |
| 36 | 17.8 | 27.9 | 24.1 | 27.0 | 27.5 | 15.7 | |
| 37 | 26.8 | 22.4 | 22.9 | 22.1 | 22.7 | 25.2 | 25.9 |
| 38 | 22.1 | 25.0 | 26.9 | 25.0 | 24.5 | 22.8 | 22.7 |
| 19-OMe | 50.6 |
Detailed 2D NMR analysis regarding this unit was thus unfolded. In 1H–1H COSY spectrum two structural fragments (C-22–C-23 and C-25–C-27–C-28) were first established by the correlations observed (Fig. 2). The connectivity of the two fragments, quaternary carbons, and three tertiary methyls was achieved by analysis of the HMBC correlations. The HMBC correlations from two tertiary methyls (CH3-30 and CH3-31) to C-23, C-28, and C-29 allowed the connections of C-23, C-28, C-30, and C-31 to the quaternary carbon C-29. Moreover, HMBC correlations from H3-26 to C-23, C-24, and C-25 linked C-23, C-25, and C-26 to C-24. Thus, a cyclocitral monoterpenoid substructure was constructed for the C10 unit. The cyclocitral unit was further linked to C-5 by HMBC correlations from H2-22 to C-4, C-5, C-6, and C-9. Although no direct HMBC correlations were available to construct the remaining ring, the still “loose end” of an oxygenated quaternary carbon (δC 82.7, C-24) and the severely downfield-shifted sp2 carbon at C-4 (δC 167.6) suggested that an oxygen bridge was located between C-24 and C-4 to form a tetrahydropyran ring. Thus, the gross structure of 1 was established, which represented an unprecedented carbon skeleton formed from coupling of a cyclocitral monoterpene and a bicyclo[3.3.1]nonane.
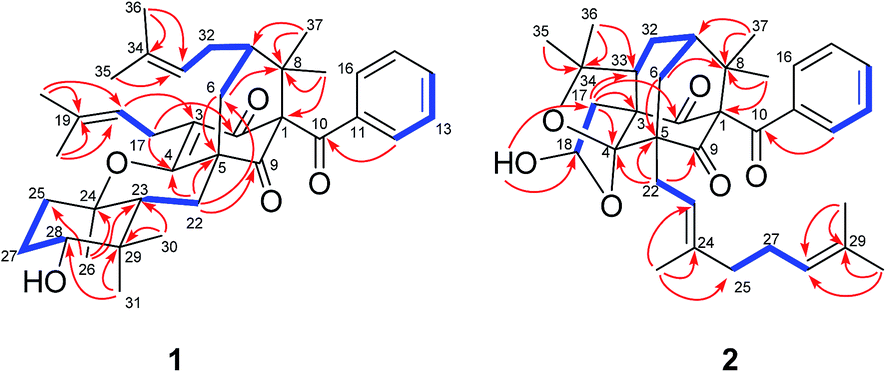 | ||
Fig. 2 Selected 1H–1H COSY ( ) and HMBC ( ) and HMBC ( ) correlations of 1 and 2. ) correlations of 1 and 2. | ||
The relative stereochemistry of 1 was established on the basis of a NOESY experiment. NOESY correlation from H3-38 to H-6a indicated that CH3-38 and H-6a adopted the axial bonds of the chair conformational D-ring and designed as β-orientation (Fig. 3). Consequently, NOESY correlations of H-6a/H-7 indicated that H-7 adopted the equatorial bond, and was assigned as β-orientation. The α-orientation of the prenyl chain at C-7 was further supported by the NOESY correlation of H-32a/H-18. In addition, NOESY correlations of H-23/H2-6, H-23/H-28, and H-28/H-25β indicated that these protons resided in the axial position of the chair conformational rings A and B, and were designated as β-oriented. Thus, the NOESY correlation of H3-31/H3-26 and H-22α indicated that CH3-26 was axially-oriented and was assigned as α-orientation.
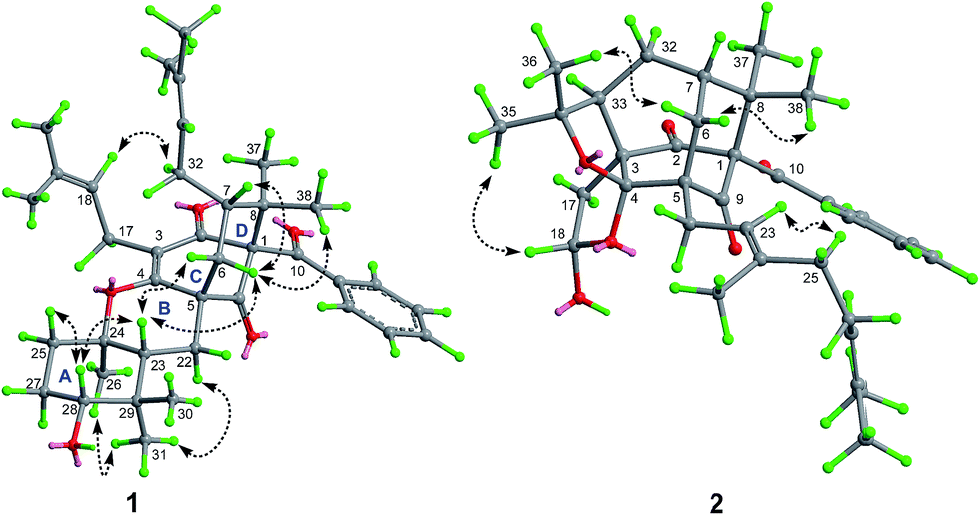 | ||
Fig. 3 Key NOE ( ) correlations of 1 and 2. ) correlations of 1 and 2. | ||
The absolute configuration of 1 was determined by quantum chemical TDDFT calculations of its theoretical ECD spectrum. Firstly, conformational analysis of the 1S,5R,7S,23R,24R,28R stereoisomer was carried out via Monte Carlo searching using molecular mechanism with MMFF94 force field in the Spartan 08 program, which resulted in 14 conformers with relative energy within 2.0 kcal mol−1.16 These conformers showed conformational possibilities on the directions of the O–H bond at C-28 (three directions) and the prenyl side chains at C-7 (two directions) and C-3 (three directions). The conformers with different directions of the O–H bond were ignored because the far distance between the OH and the chromophore had little influence on the carbon skeleton and ECD spectrum. Then six conformers (1a–1f) with different directions of the two prenyl side chains (ESI Fig. S1†) were selected for further calculation. In addition, a simplified model with the absence of two prenyl side chains (1g, ESI Fig. S1†) was also designed and calculated. As shown in Fig. 4, in the 200–400 nm region, the experimental first positive Cotton effect around 275 nm with a shoulder at 320 nm was simulated by a positive Cotton effect peak at 272 nm with a shoulder at 345 nm. The experimental negative Cotton effects around 205 nm and 242 nm were simulated correspondingly by a broad negative theoretical peak at 219 nm. Therefore, qualitative analysis of the calculated and experimental ECD spectra allowed the assignment of the absolute configuration of 1. In addition, the model conformer without the two prenyl side chains also showed similar theoretical ECD spectrum, indicating that the two side chains also had little influence on the ECD spectrum of compound 1. Thus, the absolute configurations of 1 were assigned as 1S,5R,7S,23R,24R,28R, and 1 was given a trivial name hypersampsonone A.
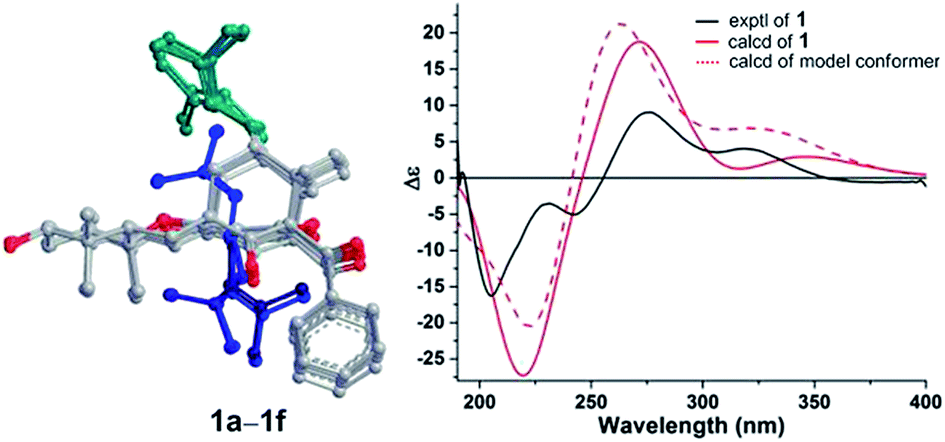 | ||
| Fig. 4 Experimental and B3LYP/6-311++G(2d,2p)//B3LYP/6-31G(d) calculated ECD spectra of 1 and the model conformer. | ||
Compound 2, a colorless oil, had the molecular formula C35H44O6, as determined by HRESIMS at m/z 559.3065 (calcd for [M − H]−, 559.3065), corresponding to 14 DOUs. The NMR data (Tables 1 and 2) and HSQC spectrum showed, in addition to a number of aliphatic methylene multiplets, signals for a benzoyl group [δH 7.36 (2H, brt, J = 7.5 Hz), 7.39 (1H, brt, J = 7.5 Hz), and 7.57 (2H, brd, J = 7.5 Hz); δC 128.3 × 2, 129.3 × 2, 132.1, 135.8, and 194.4], seven methyls [δH 1.22 (3H, s), 1.42 (3H, s), 1.47 (3H, s), 1.58 (3H, s), 1.58 (3H, s), 1.63 (3H, s), and 1.65 (3H, s); δC 16.2, 17.7, 22.4, 25.0, 25.7, 27.9, and 30.4], two keto groups [δC 207.2 and 208.1], and two trisubstituted double bonds [δH 5.07 (1H, dd, J = 6.8, 6.8 Hz) and 5.39 (1H, dd, J = 7.2, 7.2 Hz); δC 119.8, 124.3, 131.4, and 137.9]. The above-mentioned information suggested that 2 was also a PPAP derivative. Observations of characteristic signals for three sp3 quaternary carbons [δC 56.4 (C-5), δC 67.5 (C-3), and δC 115.2 (C-4)], two sp3 methines [δC 43.7 (C-7) and δC 49.4 (C-33)], two sp3 methylenes [δC 25.3 (C-32) and 31.2 (C-6)], two geminal methyls [δC 22.4 (C-37) and δC 25.0 (C-38)], and the two keto groups indicated that 2 was a homoadamantyl type PPAP with a tricyclo[4.3.1.1]undecane core.2 The geranyl and benzoyl groups were readily attached to C-5 and C-1 of the core, respectively, by HMBC correlation (Fig. 2) and 13C chemical shift analysis of C-5 and C-1.2 As 12 of the 14 DOUs were accounted for by a benzoyl group, two keto groups, two double bonds, two carbonyl groups, and the tricyclo core, the remaining two DOUs required that the rest five carbons and three oxygen atoms were constructed to give two additional rings in 2. The HMBC correlations from two tertiary methyls (CH3-35 and CH3-36) to an oxygenated quaternary carbon (δC 84.2, C-34) and C-33 indicated that a 2-oxyisopropyl was linked to C-33. The HMBC correlations from a hydroxyl proton to a hemiacetal carbon (δC 98.6) and a methylene (δC 45.8) together with those from H-17 to C-3, C-4, and C-33 linked a 2-hydroxyethyl moiety to C-3. Taking the ketal carbon of C-4 (δC 115.2) and the still “loose end” carbons of C-18 (δC 98.6) and C-34 (δC 84.2) into account, the two additional rings could only be formed, via oxygen bridges, between C-4 and C-18 and between C-4 and C-34, respectively. Thus the gross structure of 2 was figured out, which represented an unusual hexahydrofuro[2,3-b]furan-fused tricyclo[4.3.1.15,7]undecane skeleton.
The relative configuration of 2 was assigned by analyzing its NOESY data and by comparison of its 1D NMR data with those of the compounds sharing the same structural cores.2 In particular, the configurations of the two additional furan rings were readily fixed by the rigid “diamond-like” tricyclo[4.3.1.15,7] core, while the NOESY correlations of H-18/H3-35 indicated that the OH-18 at one of the furan rings was α-oriented. Compound 2 was given the trivial name hypersampsonone B.
Compound 3 had a molecular formula of C38H50O7, as established by HRESIMS. The 1H and 13C NMR spectra of 3 (Table 1 and 2) showed high similarity to those of dioxasampsone B, a known PPAP with a unique tetrahydrofuro[3,4-b]furan-fused tricyclo[4.3.1.15,7]undecane core.17 Extensive comparison of NMR data of 3 with those of dioxasampsone B revealed that their only difference arose from the side chain at C-5, where the prenyl linked to C-5 in dioxasampsone B was replaced by a geranyl in 3 to give an extended carbon skeleton for this type of compounds. Detailed 2D NMR analyses (1H–1H COSY, HSQC, and HMBC) permitted the establishment of the gross structure of 3 (ESI S25†). The relative configuration of 3 was assigned to be the same as that of dioxasampsone B by comparison of its 1D NMR and NOESY data with those of dioxasampsone B. In particular, NOE correlations of H-17/H-33 and H3-21 indicated that these protons were cofacial and designated as α-oriented. Accordingly, compound 3 was established and named hypersampsonone C.
Compound 4 displayed the HRESIMS ion at m/z 639.3683 [M + Na]+, consistent with a molecular formula of C39H52O6, 14 mass units more than that of hyphenrone N (8),2 a co-isolated metabolite in the current study. The 1H and 13C NMR data of 4 (Tables 1 and 2) were very similar to those of 8 except for the presence of an additional methoxyl (δH 2.79) and the downfield-shifted carbon signal at C-19 [δC 75.2 in 4, δC 71.4 in 8], indicating that 4 was a 19-O-methyl derivative of 8. This was further confirmed by HMBC correlation from the methoxyl to C-19. The relative configuration of 4 was assigned to be the same as that of 8 by comparing their 1D NMR data and by analyzing its NOESY data. Thus, compound 4 was deduced as depicted and was given the trivial name hypersampsonone D.
Compound 5, a colorless oil, had the molecular formula C35H44O6 as determined by HRESIMS at m/z 561.3225 (calcd 561.3211). The 1D NMR data of 5 was very similar to that of hypersampsone N (9),18 except for the replacement of the prenyl group in 9 by a geranyl group in 5. Detailed 2D NMR analyses (1H–1H COSY, HSQC, and HMBC) permitted the establishment of the gross structure of 5 as depicted. The relative configuration of 5 was assigned to be the same as that of 9 by NOESY experiment and by comparison of their NMR data. Thus, compound 5 was deduced as shown and was given the trivial name hypersampsonone E.
The HRESIMS of 6 gave a pseudo-molecular ion at m/z 601.3511 [M − H]− (calcd 601.3535), corresponding to the molecular formula of C38H50O6, with one more O-atom than that of sampsonione C (10).19 The 13C NMR data of 6 was very similar to that of 10, with the differences scattering around C-19. In comparison with 10, the carbon resonances of C-18, C-19, C-20, and C-21 in 6 were shifted by ΔδC −3.1, +12.2, −5.3, −7.1 ppm, respectively, indicating that 6 possessed a hydroperoxy group at C-19.20 The structure of 6 was further confirmed by the conversion of 6 to 10 in acidic CDCl3 under several days' storage. Accordingly, compound 6 was established as depicted and was named hypersampsonone F.
Compound 7, a colorless oil, had the molecular formula C38H50O5 as determined by HRESIMS at m/z 587.3758 [M + H]+ (calcd 587.3731), indicating that it was an isomer of sampsonione C (10).19 Comprehensive analysis of 1D and 2D NMR spectra revealed that 7 shared the same planar structure with 10. The only structural difference between 7 and 10 was occurring at C-18, as implied by the slightly shifted carbon resonances around this stereocenter (ΔC-17, ΔC-18, and ΔC-34 within 2 ppm), indicating that 7 was a 18-epimer of 10. This was further supported by the NOE correlations of H3-35/H-18 and H-33. Thus, compound 7 was assigned as depicted and was named hypersampsonone G.
The absolute stereochemistry of 2–7 were assigned by CD analysis. Based on the CD benchmark summarized by Zhang et al.,11 the R configuration for C-1 of benzoyl substituted adamantyl and homoadamantyl PPAPs would generate a negative Cotton effect around 333 nm. Thus, compounds 2–5 (homoadamantyl type) and 6–7 (adamantane type) displaying the negative cotton effects around 330 nm (ESI S3†) were assigned the R configuration at C-1. This assignment was consistent with the biosynthetic origin of the PPAPs from the genus Hypericum.11,17 Hence, The absolute configuration of 2–7 was assigned as depicted.
The known compounds hyphenrone N (8),2 hypersampsone N (9),18 sampsonione C (10),19 hypersampsone Q (11),18 plukenetione B (12),21 hyperattenin I (13),22 sampsonione F (14),19 attenuatumione D (15),23 sampsonione G (16),19 hypercohone A (17),21 otogirinin D (18),24 hyperattenin C (19),22 hyperibone A (20),25 hyperattenin G (21),22 cowabenzophenone B (22),26 sampsonione H (23),19 hypersampsone P (24),18 sampsonione A (25),27 sampsonione B (26),27 hypersampsone N (27),18 sampsonione L (28),28 hyperisampsin I (29),3 and hyperisampsin J (30)3 were identified by comparison of their NMR and MS data with those in the literature.
Most of the structurally related PPAPs isolated from the genus Hypericum are believed to be biosynthesized from the biogenetically acceptable 2,4,6-trihydroxybenzophenone i.2,17,28 It is further decorated with dimethylallyl diphosphates (DMAPP) to give intermediate ii, which is the common precursor of diverse PPAPs.2,17,28 A plausible biosynthetic pathway of 1–30 starting from ii through a series of reactions including epoxidation, intramolecular cyclization, oxidation cleavage, dehydration, reduction, and cationic polycyclization17,28,29 was proposed in Scheme 1.
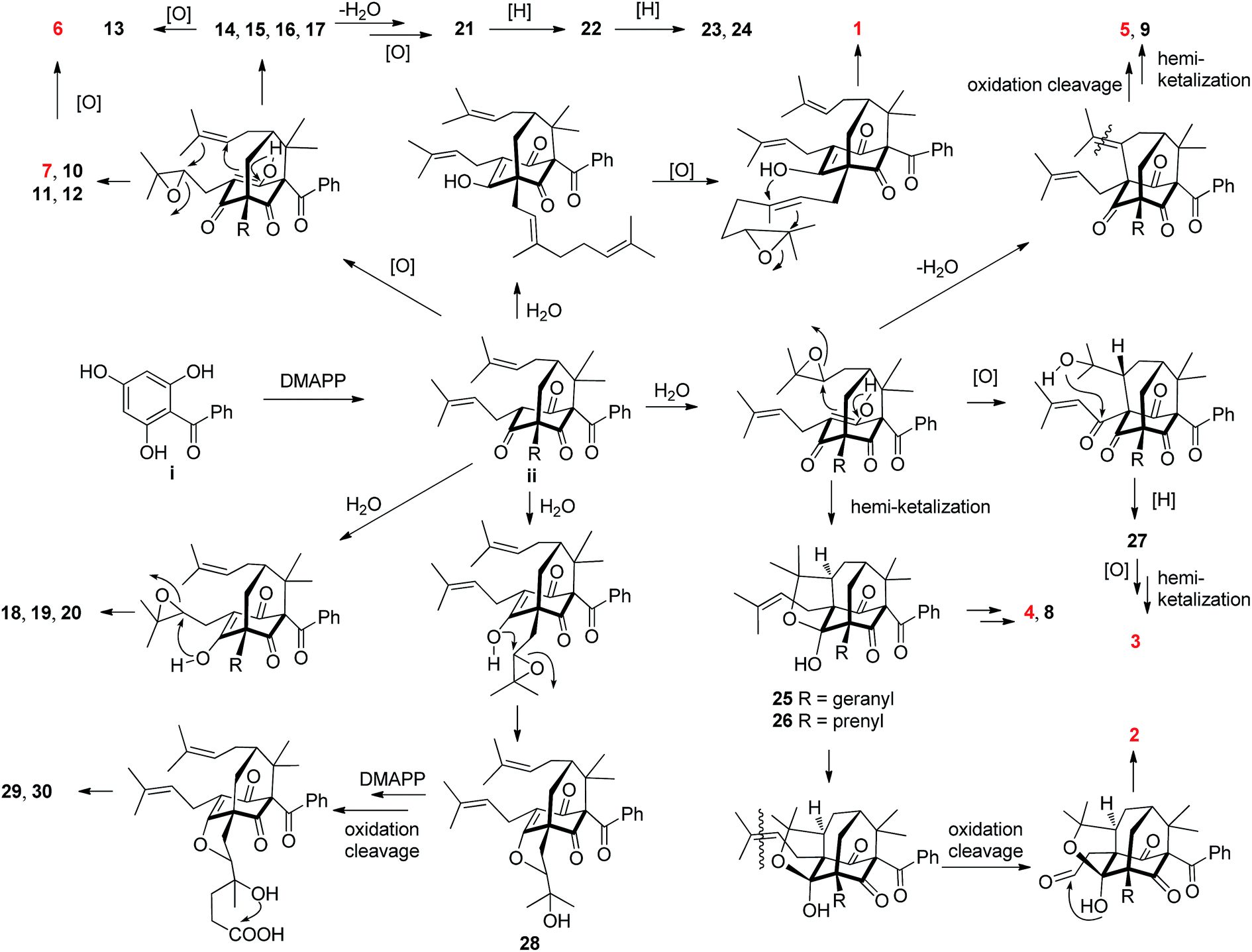 | ||
| Scheme 1 Plausible biosynthetic pathway for 1–30. | ||
Compounds 1–30 were tested for their inhibitory activity against PED4D2 at an initial concentration of 10 μM by using tritium-labeled adenosine 3′,5′-cyclic monophosphate ([3H]-cAMP) as substrate (ESI S1†). Rolipram, a well-known PDE4 inhibitor, was used as the reference compound (IC50 = 0.62 μM), comparable to the reported value of 1.0 μM.30 Compounds with inhibition greater than 50% at 10 μM were further analyzed to determine their corresponding IC50 values. As shown in Table 3, compounds 1, 18–19, 21 and 25–30 had significant inhibitory activity against PDE4D2, with IC50 values less than 10 μM, and 19 turned out to be the most active compound with an IC50 value of 0.64 μM, being comparable to that of rolipram. The inhibitory curves of compounds 19, 27, 30, and rolipram are represented in Fig. 5.
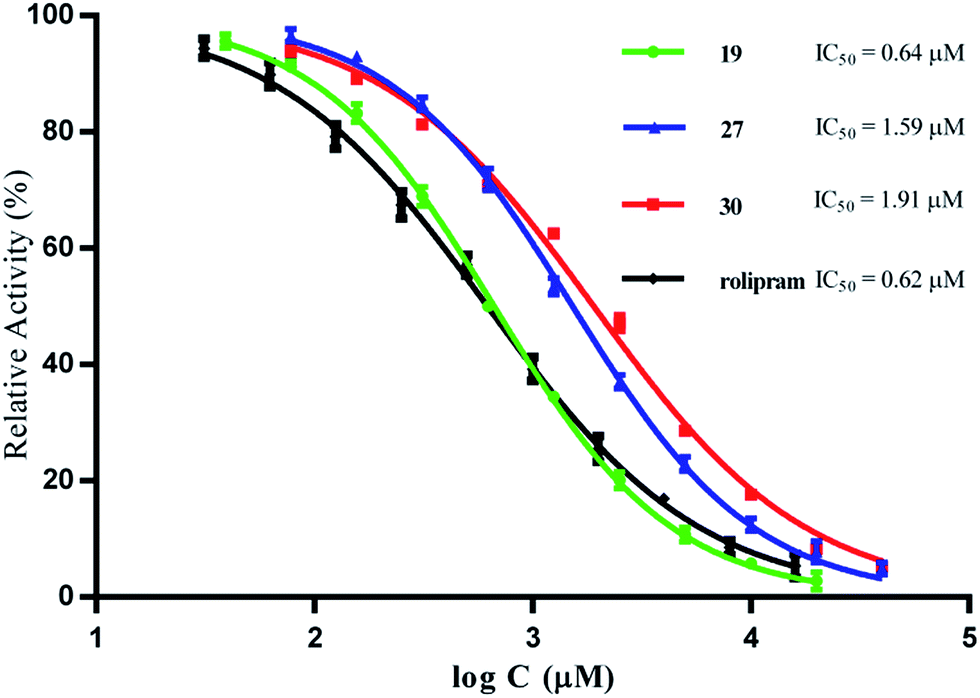 | ||
| Fig. 5 Inhibitory curves of compounds 19, 27, 30 and rolipram (positive control) against PDE4D2. | ||
Conclusions
In summary, seven new polycyclic polyprenylated acylphloroglucinols (1–7) and 23 known analogs (8–30) were isolated from aerial parts of Hypericum sampsonii. Compound 1 represents an unprecedented cyclocitral monoterpene-coupled bicyclo[3.3.1]nonane skeleton, while 2 features an unusual hexahydrofuro[2,3-b]furan-diepoxy ring system fused in a tricyclo[4.3.1.15,7]undecane skeleton. Ten compounds exhibited inhibitory activity against PDE4D2 with IC50 values less than 10 μM, and the most active compound, 19 (IC50 = 0.64 μM), was comparable to that of rolipram (IC50 = 0.62 μM).Natural PDE4 inhibitors are rare, and the current study revealed a new group of PDE4 inhibitors from Hypericum sampsonii, which not only expand the structural diversity of PPAPs categories, but also explain the anti-inflammatory efficacy of this plant in traditional Chinese medicine. Studies toward their selectivity versus other PDE members are in progress.
Experimental section
General experimental procedures
Optical rotations were measured on a Perkin-Elmer 341 polarimeter. UV spectra were recorded on a Shimadzu UV-2450 spectrophotometer. IR spectra were determined on a Bruker Tensor 37 infrared spectrophotometer with KBr disks. NMR spectra were measured on a Bruker AM-400 spectrometer at 25 °C. ESIMS and HRESIMS were carried out on a Finnigan LCQ Deca instrument. A Shimadzu LC-20AT equipped with a SPD-M20A PDA detector was used for HPLC, and a YMC-pack ODS-A column (250 × 10 mm, S-5 μm, 12 nm) was used for semipreparative HPLC separation. A chiral column (Phenomenex Lux, cellulose-2, 250 × 10 mm, 5 μm) was used for chiral separation. Silica gel (300–400 mesh, Qingdao Haiyang Chemical Co. Ltd.), reversed-phase C18 (RP-C18) silica gel (12 nm, S-50 μm, YMC Co. Ltd.), and MCI gel (CHP20P, 75–150 μm, Mitsubishi Chemical Industries Ltd.) were used for column chromatography (CC). All solvents were of analytical grade (Guangzhou Chemical Reagents Company, Ltd.).Plant material
The aerial parts of H. sampsonii were collected in August from Jiangsu province, and were identified by Dr Gui-Hua Tang of Sun Yat-sen University. A voucher specimen (accession number: YBC201411) has been deposited at the School of Pharmaceutical Sciences, Sun Yat-sen University.Extraction and isolation
The air-dried powder of the aerial parts of H. sampsonii (5 kg) was extracted with 95% EtOH (3 × 5 L) at room temperature to give 450 g of crude extract. The extract was suspended in H2O (3 L) and successively partitioned with petroleum ether (PE, 3 × 3 L) and EtOAc (3 × 3 L). The petroleum ether extract (181 g) was subjected to MCI gel column chromatography (CC) eluted with a MeOH/H2O gradient (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 → 10:0) to afford five fractions (I–V). Fraction II (22.6 g) was chromatographed over a silica gel CC (n-hexane/EtOAc, 40:1) to give four fractions (Fr. IIa–IId). Fr. IIb (2.9 g) was subjected to C18 reversed-phase (RP-C18) silica gel CC eluted with MeOH/H2O (8:2 → 10:0) to afford four fractions (Fr. IIb1–IIb4). Fr. IIb1 (162 mg) was further purified on RP-C18 HPLC using MeOH/H2O (90:10, 3 mL min−1) as mobile phase to give 19 (35 mg, tR 18.5 min), 18 (15 mg, tR 19.5 min), and 9 (8 mg, tR 22 min), Fr. IIb2 (40 mg) further purified on RP-C18 HPLC using MeCN/H2O (80:20, 3 mL min−1) as mobile phase to afford 1 (11 mg, tR 8.5 min), 28 (7 mg, tR 12.5 min) and 5 (3.4 mg, tR 15.5 min). Fr. IIb3 (362 mg) was further purified on RP-C18 HPLC using MeOH/H2O (90:10, 3 mL min−1) as mobile phase to give 7 (50 mg, tR 18 min), 14 (15 mg, tR 18.5 min), 10 (25 mg, tR 19.5 min), and 15 (16 mg, tR 21 min). Fr. IIb4 (145 mg) was further purified on RP-C18 HPLC using MeCN/H2O (80:20, 3 mL min−1) as mobile phase to give 17 (24 mg, tR 14 min), 11 (15 mg, tR 16.5 min), 16 (11 mg, tR 17.5 min), and 12 (16 mg, tR 21 min). Fraction III (12.6 g) was chromatographed over a silica gel CC (n-hexane/EtOAc, 50:1) to give five fractions (Fr. IIIa–IIIe). Fr. IIIb (1.9 g) was subjected to C18 reversed-phase (RP-C18) silica gel CC eluted with MeOH/H2O (6:4 → 10:0) and further purified on RP-C18 HPLC using MeOH/H2O (90:10, 3 mL min−1) as mobile phase to give 4 (8.4 mg, tR 21 min), 29 (4 mg, tR 24 min) and 30 (6.4 mg, tR 24.5 min). Fr. IIIc (45 mg) was further purified on RP-C18 HPLC using MeCN/H2O (85:15, 3 mL min−1) as mobile phase to give 13 (8 mg, tR 14 min) and 6 (6 mg, tR 16.5 min). Fr. IIId (370 mg) was chromatographed over a Sephadex LH-20 eluted with MeOH, and further purified on RP-C18 HPLC using MeOH/H2O (90:10, 3 mL min−1) as mobile phase to afford 2 (5.6 mg, tR 13 min), 25 (12.6 mg, tR 16 min), and 26 (3.8 mg, tR 18 min). Fr. IIIe (1.9 g) was chromatographed over a Sephadex LH-20 eluted with MeOH to give two fractions (Fr. IIIe1–IIIe2). Fr. IIIe1 (36 mg) was further purified on RP-C18 HPLC using MeCN/H2O (80:20, 3 mL min−1) as mobile phase to give 22 (14 mg, tR 12.4 min) and 20 (6 mg, tR 16.5 min), while Fr. IIIe2 (900 mg) followed by a C18 reversed-phase (RP-C18) silica gel CC eluted with MeOH/H2O (8:2 → 10:0) to afford 3 (6.2 mg) and 8 (8.8 mg). Fraction IV (8 g) was chromatographed over a Sephadex LH-20 (CH2Cl2/MeOH, v/v, 1:1), to give three fractions (Fr. IVa–IVc). Fr. IVa was further purified on RP-C18 HPLC using MeCN/H2O (90:10, 3 mL min−1) as mobile phase to give 23 (6 mg, tR 14 min), 24 (5 mg, tR 16 min), 21 (9 mg, tR 21 min) and 27 (6 mg, tR 21.5 min).
ε) 206.5 (7.10), 247 (6.89), 286 (6.85) nm; IR (KBr) νmax 3505, 2925, 1721, 1694, 1633, 1585, 1221, 745 cm−1; CD (c 2.85 × 10−4 M, CH3CN) λmax (Δε) 205 (−16.31), 242 (−5.06), 275 (+9.04), 318 (+4.02) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 585.3561 (calcd for C38H49O5− [M − H]−, 585.3585).ε) 208.5 (7.51), 247.5 (7.38) nm; IR (KBr) νmax 3442, 2927, 1725, 1691, 1450, 1224 cm−1; CD (c 6.21 × 10−4 M, CH3CN) λmax (Δε) 216 (+1.86), 243 (+7.20), 258 (+1.45), 297 (−7.09) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 559.3065 (calcd for C35H43O6− [M − H]−, 559.3065).ε) 207 (7.14), 246.5 (6.83) nm; IR (KBr) νmax 3443, 2927, 1724, 1693, 1227 cm−1; CD (c 4.09 × 10−4 M, CH3CN) λmax (Δε) 213 (+15.06), 243 (−6.57), 259 (−6.58), 283 (+3.30), 314 (−2.31) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 641.3480 (calcd for C38H50O7Na+ [M + Na]+, 641.3449).ε) 208.5 (7.27), 245.5 (7.07) nm; IR (KBr) νmax 2979, 1694, 1223, 764, 749 cm−1; CD (c 2.33 × 10−4 M, CH3CN) λmax (Δε) 243 (+3.46), 297 (−3.34) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 639.3683 (calcd for C39H52O6Na+ [M + Na]+, 639.3656).ε) 205 (3.64), 244 (3.18) nm; IR (KBr) νmax 2927, 1738, 1703, 1597, 1236 cm−1; CD (c 2.33 × 10−4 M, CH3CN) λmax (Δε) 206 (−15.51), 211 (+5.49), 251 (+4.57), 284 (−1.36), 322 (−2.06) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 561.3225 (calcd for C35H45O6+ [M + H]+, 561.3211).ε) 207.5 (7.32), 245.5 (7.12) nm; IR (KBr) νmax 2979, 1694, 1223, 764, 749 cm−1; CD (c 2.45 × 10−4 M, CH3CN) λmax (Δε) 208 (+9.21), 238 (−6.11), 258 (+1.45), 311 (−3.02), 324 (−2.96) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 601.3511 (calcd for C38H49O6− [M − H]−, 601.3535).ε) 208 (4.49), 245 (4.31) nm; IR (KBr) νmax 2971, 2935, 1734, 1696, 1222 cm−1; CD (c 4.55 × 10−4 M, CH3CN) λmax (Δε) 218 (+4.15), 241 (−7.37), 285 (+2.11), 324 (−2.61) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 587.3758 (calcd for C38H51O5+ [M + H]+, 587.3731).
:7 → 10:0) to afford five fractions (I–V). Fraction II (22.6 g) was chromatographed over a silica gel CC (n-hexane/EtOAc, 40:1) to give four fractions (Fr. IIa–IId). Fr. IIb (2.9 g) was subjected to C18 reversed-phase (RP-C18) silica gel CC eluted with MeOH/H2O (8:2 → 10:0) to afford four fractions (Fr. IIb1–IIb4). Fr. IIb1 (162 mg) was further purified on RP-C18 HPLC using MeOH/H2O (90:10, 3 mL min−1) as mobile phase to give 19 (35 mg, tR 18.5 min), 18 (15 mg, tR 19.5 min), and 9 (8 mg, tR 22 min), Fr. IIb2 (40 mg) further purified on RP-C18 HPLC using MeCN/H2O (80:20, 3 mL min−1) as mobile phase to afford 1 (11 mg, tR 8.5 min), 28 (7 mg, tR 12.5 min) and 5 (3.4 mg, tR 15.5 min). Fr. IIb3 (362 mg) was further purified on RP-C18 HPLC using MeOH/H2O (90:10, 3 mL min−1) as mobile phase to give 7 (50 mg, tR 18 min), 14 (15 mg, tR 18.5 min), 10 (25 mg, tR 19.5 min), and 15 (16 mg, tR 21 min). Fr. IIb4 (145 mg) was further purified on RP-C18 HPLC using MeCN/H2O (80:20, 3 mL min−1) as mobile phase to give 17 (24 mg, tR 14 min), 11 (15 mg, tR 16.5 min), 16 (11 mg, tR 17.5 min), and 12 (16 mg, tR 21 min). Fraction III (12.6 g) was chromatographed over a silica gel CC (n-hexane/EtOAc, 50:1) to give five fractions (Fr. IIIa–IIIe). Fr. IIIb (1.9 g) was subjected to C18 reversed-phase (RP-C18) silica gel CC eluted with MeOH/H2O (6:4 → 10:0) and further purified on RP-C18 HPLC using MeOH/H2O (90:10, 3 mL min−1) as mobile phase to give 4 (8.4 mg, tR 21 min), 29 (4 mg, tR 24 min) and 30 (6.4 mg, tR 24.5 min). Fr. IIIc (45 mg) was further purified on RP-C18 HPLC using MeCN/H2O (85:15, 3 mL min−1) as mobile phase to give 13 (8 mg, tR 14 min) and 6 (6 mg, tR 16.5 min). Fr. IIId (370 mg) was chromatographed over a Sephadex LH-20 eluted with MeOH, and further purified on RP-C18 HPLC using MeOH/H2O (90:10, 3 mL min−1) as mobile phase to afford 2 (5.6 mg, tR 13 min), 25 (12.6 mg, tR 16 min), and 26 (3.8 mg, tR 18 min). Fr. IIIe (1.9 g) was chromatographed over a Sephadex LH-20 eluted with MeOH to give two fractions (Fr. IIIe1–IIIe2). Fr. IIIe1 (36 mg) was further purified on RP-C18 HPLC using MeCN/H2O (80:20, 3 mL min−1) as mobile phase to give 22 (14 mg, tR 12.4 min) and 20 (6 mg, tR 16.5 min), while Fr. IIIe2 (900 mg) followed by a C18 reversed-phase (RP-C18) silica gel CC eluted with MeOH/H2O (8:2 → 10:0) to afford 3 (6.2 mg) and 8 (8.8 mg). Fraction IV (8 g) was chromatographed over a Sephadex LH-20 (CH2Cl2/MeOH, v/v, 1:1), to give three fractions (Fr. IVa–IVc). Fr. IVa was further purified on RP-C18 HPLC using MeCN/H2O (90:10, 3 mL min−1) as mobile phase to give 23 (6 mg, tR 14 min), 24 (5 mg, tR 16 min), 21 (9 mg, tR 21 min) and 27 (6 mg, tR 21.5 min).
ε) 206.5 (7.10), 247 (6.89), 286 (6.85) nm; IR (KBr) νmax 3505, 2925, 1721, 1694, 1633, 1585, 1221, 745 cm−1; CD (c 2.85 × 10−4 M, CH3CN) λmax (Δε) 205 (−16.31), 242 (−5.06), 275 (+9.04), 318 (+4.02) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 585.3561 (calcd for C38H49O5− [M − H]−, 585.3585).ε) 208.5 (7.51), 247.5 (7.38) nm; IR (KBr) νmax 3442, 2927, 1725, 1691, 1450, 1224 cm−1; CD (c 6.21 × 10−4 M, CH3CN) λmax (Δε) 216 (+1.86), 243 (+7.20), 258 (+1.45), 297 (−7.09) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 559.3065 (calcd for C35H43O6− [M − H]−, 559.3065).ε) 207 (7.14), 246.5 (6.83) nm; IR (KBr) νmax 3443, 2927, 1724, 1693, 1227 cm−1; CD (c 4.09 × 10−4 M, CH3CN) λmax (Δε) 213 (+15.06), 243 (−6.57), 259 (−6.58), 283 (+3.30), 314 (−2.31) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 641.3480 (calcd for C38H50O7Na+ [M + Na]+, 641.3449).ε) 208.5 (7.27), 245.5 (7.07) nm; IR (KBr) νmax 2979, 1694, 1223, 764, 749 cm−1; CD (c 2.33 × 10−4 M, CH3CN) λmax (Δε) 243 (+3.46), 297 (−3.34) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 639.3683 (calcd for C39H52O6Na+ [M + Na]+, 639.3656).ε) 205 (3.64), 244 (3.18) nm; IR (KBr) νmax 2927, 1738, 1703, 1597, 1236 cm−1; CD (c 2.33 × 10−4 M, CH3CN) λmax (Δε) 206 (−15.51), 211 (+5.49), 251 (+4.57), 284 (−1.36), 322 (−2.06) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 561.3225 (calcd for C35H45O6+ [M + H]+, 561.3211).ε) 207.5 (7.32), 245.5 (7.12) nm; IR (KBr) νmax 2979, 1694, 1223, 764, 749 cm−1; CD (c 2.45 × 10−4 M, CH3CN) λmax (Δε) 208 (+9.21), 238 (−6.11), 258 (+1.45), 311 (−3.02), 324 (−2.96) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 601.3511 (calcd for C38H49O6− [M − H]−, 601.3535).ε) 208 (4.49), 245 (4.31) nm; IR (KBr) νmax 2971, 2935, 1734, 1696, 1222 cm−1; CD (c 4.55 × 10−4 M, CH3CN) λmax (Δε) 218 (+4.15), 241 (−7.37), 285 (+2.11), 324 (−2.61) nm; 1H and 13C NMR data see Tables 1 and 2; HRESIMS m/z 587.3758 (calcd for C38H51O5+ [M + H]+, 587.3731).Acknowledgements
The authors thank the National High Technology Research and Development Program of China (863 Project, No. 2015AA020928), the Guangdong Natural Science Funds for Distinguished Young Scholar (No. 2014A030306047), and the National Natural Science Foundation of China (No. 81573302 and No. 81402813) for providing financial support to this work.Notes and references
- R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963–3986 CrossRef CAS PubMed.
- X. W. Yang, M. M. Li, X. Liu, D. Ferreira, Y. Ding, J. J. Zhang, Y. Liao, H. B. Qin and G. Xu, J. Nat. Prod., 2015, 78, 885–895 CrossRef CAS PubMed.
- H. C. Zhu, C. M. Chen, Q. Y. Tong, X. T. Chen, J. Yang, J. J. Liu, B. Sun, J. P. Wang, G. M. Yao, Z. W. Luo, Y. B. Xue and Y. H. Zhang, Sci. Rep., 2015, 5, 14772 CrossRef CAS PubMed.
- J. J. Zhang, J. Yang, Y. Liao, X. W. Yang, J. Z. Ma, Q. L. Xiao, L. X. Yang and G. Xu, Org. Lett., 2014, 16, 4912–4915 CrossRef CAS PubMed.
- W. J. Tian, Y. Yu, X. J. Yao, H. F. Chen, Y. Dai, X. K. Zhang and X. S. Yao, Org. Lett., 2014, 16, 3448–3451 CrossRef CAS PubMed.
- B. A. Sparling, J. K. Tucker, D. C. Moebius and M. D. Shair, Org. Lett., 2015, 17, 3398–3401 CrossRef CAS PubMed.
- J. A. Richard, Eur. J. Org. Chem., 2014, 2014, 273–299 CrossRef CAS.
- C. P. Ting and T. J. Maimone, J. Am. Chem. Soc., 2015, 137, 10516–10519 CAS.
- Jiangsu New Medical College, Dictionary of Chinese Crude Drugs, Shanghai Scientific Technological Publishers, Shanghai, 1977, pp. 345–346 Search PubMed.
- Z. Y. Xiao, Q. Mu, W. K. P. Shiu, Y. H. Zeng and S. Gibbons, J. Nat. Prod., 2007, 70, 1779–1782 CrossRef CAS PubMed.
- H. C. Zhu, C. M. Chen, J. Yang, X. N. Li, J. J. Liu, B. Sun, S. X. Huang, D. Y. Li, G. M. Yao, Z. W. Luo, Y. Li, J. W. Zhang, Y. B. Xue and Y. H. Zhang, Org. Lett., 2014, 16, 6322–6325 CrossRef CAS PubMed.
- A. B. Burgin, O. T. Magnusson, J. Singh, P. Witte, B. L. Staker, J. M. Bjornsson, M. Thorsteinsdottir, S. Hrafnsdottir, T. Hagen, A. S. Kiselyov, L. J. Stewart and M. E. Gurney, Nat. Biotechnol., 2010, 28, 63–70 CrossRef CAS PubMed.
- T. T. Lin, Y. Y. Huang, G. H. Tang, Z. B. Cheng, X. Liu, H. B. Luo and S. Yin, J. Nat. Prod., 2014, 77, 955–962 CrossRef CAS PubMed.
- X. Liu, H. B. Luo, Y. Y. Huang, J. M. Bao, G. H. Tang, Y. Y. Chen, J. Wang and S. Yin, Org. Lett., 2015, 16, 282–285 CrossRef PubMed.
- I. M. Hernandez, M. C. Fernandez, O. Cuesta-Rubio, A. L. Piccinelli and L. Rastrelli, J. Nat. Prod., 2005, 68, 931–934 CrossRef PubMed.
- Spartan 08, Wavefunction Inc., Irvine, CA Search PubMed.
- W. J. Tian, Y. Q. Qiu, X. J. Yao, H. F. Chen, Y. Dai, X. K. Zhang and X. S. Yao, Org. Lett., 2014, 16, 6346–6349 CrossRef CAS PubMed.
- W. J. Tian, Y. Q. Qiu, X. J. Jin, H. F. Chen, X. J. Yao, Y. Dai and X. S. Yao, Tetrahedron, 2014, 70, 7912–7916 CrossRef CAS.
- L. H. Hu and K. Y. Sim, Tetrahedron Lett., 1999, 40, 759–762 CrossRef CAS.
- S. Yin, X. Chen, Z. S. Su, S. P. Yang, C. Q. Fan, J. Ding and J. M. Yue, Tetrahedron, 2009, 65, 1147–1152 CrossRef CAS.
- X. Liu, X. W. Yang, C. Q. Chen, C. Y. Wu, J. J. Zhang, J. Z. Ma, H. Wang, Q. S. Zhao, L. X. Yang and G. Xu, Nat. Prod. Bioprospect., 2013, 3, 233–237 CrossRef CAS.
- D. Y. Li, Y. B. Xue, H. C. Zhu, Y. Li, B. Sun, J. J. Liu, G. M. Yao, J. W. Zhang, G. Du and Y. H. Zhang, RSC Adv., 2015, 5, 5277–5287 RSC.
- Z. B. Zhou, Y. M. Zhang, K. Pan, J. G. Luo and L. Y. Kong, Fitoterapia, 2014, 95, 1–7 CrossRef CAS PubMed.
- Y. Ishida, O. Shirota, S. Sekita, K. Someya, F. Tokita, T. Nakane and M. Kuroyanagi, Chem. Pharm. Bull., 2010, 58, 336–343 CrossRef CAS PubMed.
- M. Matsuhisa, Y. Shikishima, Y. Takaishi, G. Honda, M. Ito, Y. Takeda, H. Shibata, T. Higuti, O. K. Kodzhimatov and O. Ashurmetov, J. Nat. Prod., 2002, 65, 290–294 CrossRef CAS.
- T. Sriyatep, W. Maneerat, T. Sripisut, S. Cheenpracha, T. Machan, W. Phakhodee and S. Laphookhieo, Fitoterapia, 2014, 92, 285–289 CrossRef CAS PubMed.
- L. H. Hu and K. Y. Sim, Tetrahedron Lett., 1998, 39, 7999–8002 CrossRef CAS.
- L. H. Hu and K. Y. Sim, Tetrahedron, 2000, 56, 1379–1386 CrossRef CAS.
- G. Rajendar and E. J. Corey, J. Am. Chem. Soc., 2015, 137, 5837–5844 CrossRef CAS PubMed.
- A. T. Bender and J. A. Beavo, Pharmacol. Rev., 2006, 58, 488–520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: HRESIMS, 1D and 2D NMR spectra of 1–7. See DOI: 10.1039/c6ra08805h |
| This journal is © The Royal Society of Chemistry 2016 |